Structure factor
In condensed matter physics and crystallography, the static structure factor (or structure factor for short) is a mathematical description of how a material scatters incident radiation. The structure factor is a particularly useful tool in the interpretation of interference patterns obtained in X-ray, electron and neutron diffraction experiments.
The static structure factor is measured without resolving the energy of scattered photons/electrons/neutrons. Energy-resolved measurements yield the dynamic structure factor.
Derivation
Let us consider a scalar (real) quantity 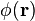 defined in a volume
defined in a volume  ; it may correspond, for instance, to a mass or charge distribution or to the refractive index of an inhomogeneous medium. If the scalar function is assumed to be integrable, we can define its Fourier transform
; it may correspond, for instance, to a mass or charge distribution or to the refractive index of an inhomogeneous medium. If the scalar function is assumed to be integrable, we can define its Fourier transform  . Expressing the field
. Expressing the field  in terms of the spatial frequency
in terms of the spatial frequency  instead of the point position
instead of the point position  is very useful, for instance, when interpreting scattering experiments. Indeed, in the Born approximation (weak interaction between the field and the medium), the amplitude of the signal corresponding to the scattering vector is proportional to
is very useful, for instance, when interpreting scattering experiments. Indeed, in the Born approximation (weak interaction between the field and the medium), the amplitude of the signal corresponding to the scattering vector is proportional to 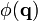 . Very often, only the intensity of the scattered signal
. Very often, only the intensity of the scattered signal 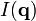 is detectable, so that
is detectable, so that  .
.
If the system under study is composed of a number  of identical constituents (atoms, molecules, colloidal particles, etc.) it is very convenient to explicitly capture the variation in due to the morphology of the individual particles using an auxiliary function
of identical constituents (atoms, molecules, colloidal particles, etc.) it is very convenient to explicitly capture the variation in due to the morphology of the individual particles using an auxiliary function  , such that:
, such that:
-
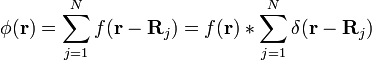 ,
,(1)
with  the particle positions. In the second equality, the field is decomposed as the convolution product
the particle positions. In the second equality, the field is decomposed as the convolution product 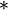 of the function
of the function  , describing the "form" of the particles, with a sum of Dirac delta functions depending only on their positions. Using the property that the Fourier transform of a convolution product is simply the product of the Fourier transforms of the two factors, we have
, describing the "form" of the particles, with a sum of Dirac delta functions depending only on their positions. Using the property that the Fourier transform of a convolution product is simply the product of the Fourier transforms of the two factors, we have 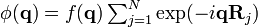 , such that:
, such that:
-
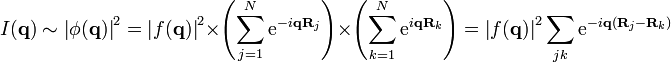 .
.(2)
In general, the particle positions are not fixed and the measurement takes place over a finite exposure time and with a macroscopic sample (much larger than the interparticle distance). The experimentally accessible intensity is thus an averaged one  ; we need not specify whether
; we need not specify whether  denotes a time or ensemble average. We can finally write:
denotes a time or ensemble average. We can finally write:
-
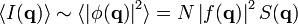 ,
,(3)
thus defining the structure factor
-
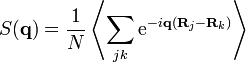 .
.(4)
Perfect crystals
In a crystal, the constitutive particles are arranged periodically, forming a lattice. In the following, we will consider that all particles are identical (so the above separation in factor and structure factors (3) holds). We also assume that all atoms have an identical environment (i.e. they form a Bravais lattice). The general case of lattice with a basis (see below) is not fundamentally different.
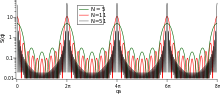
If the lattice is infinite and completely regular, the system is a perfect crystal. In addition, we will neglect all thermal motion, so that there is no need for averaging in (4). As in (2), we can write:
-
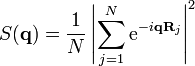 .
.
The structure factor is simply the squared modulus of the Fourier transform of the lattice, and it is itself a periodic arrangement of points, known as the reciprocal lattice.
One dimension
 .
.The reciprocal lattice is easily constructed in one dimension: for particles on a line with a period  , the atom positions
, the atom positions 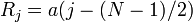 (for simplicity, we consider that is odd). The sum of the phase factors is a simple geometric series and the structure factor becomes:
(for simplicity, we consider that is odd). The sum of the phase factors is a simple geometric series and the structure factor becomes:
-
![S(q)={\frac {1}{N}}\left|{\frac {1-{\mathrm {e}}^{{-iNqa}}}{1-{\mathrm {e}}^{{-iqa}}}}\right|^{2}={\frac {1}{N}}\left[{\frac {\sin(Nqa/2)}{\sin(qa/2)}}\right]^{2}](/2014-wikipedia_en_all_02_2014/I/media/d/9/e/5/d9e58be7b9a0c212f87d7cbea54d6eea.png) .
.
This function is shown in the Figure below for different values of .
Based on this expression for  , one can draw several conclusions: the reciprocal lattice has a spacing
, one can draw several conclusions: the reciprocal lattice has a spacing 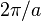 ; the intensity of the maxima increases with the number of particles
; the intensity of the maxima increases with the number of particles  (this is apparent from the Figure and can be shown by estimating the limit
(this is apparent from the Figure and can be shown by estimating the limit  using, for instance, L'Hôpital's rule); the intensity at the midpoint
using, for instance, L'Hôpital's rule); the intensity at the midpoint  (by direct evaluation); the peak width also decreases like
(by direct evaluation); the peak width also decreases like 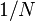 . In the large limit, the peaks become infinitely sharp Dirac delta functions.
. In the large limit, the peaks become infinitely sharp Dirac delta functions.
Two dimensions
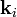 ,
,  and the scattering vector .
and the scattering vector .In two dimensions, there are only five Bravais lattices. The corresponding reciprocal lattices have the same symmetry as the direct lattice. The Figure shows the construction of one vector of the reciprocal lattice and its relation with a scattering experiment.
A parallel beam, with wave vector is incident on a square lattice of parameter . The scattered wave is detected at a certain angle, which defines the wave vector of the outgoing beam, (under the assumption of elastic scattering, 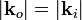 ). One can equally define the scattering vector
). One can equally define the scattering vector  and construct the harmonic pattern
and construct the harmonic pattern 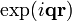 . In the depicted example, the spacing of this pattern coincides to the distance between particle rows:
. In the depicted example, the spacing of this pattern coincides to the distance between particle rows: 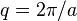 , so that contributions to the scattering from all particles are in phase (constructive interference). Thus, the total signal in direction is strong, and belongs to the reciprocal lattice. It is easily shown that this configuration fulfills Bragg's law.
, so that contributions to the scattering from all particles are in phase (constructive interference). Thus, the total signal in direction is strong, and belongs to the reciprocal lattice. It is easily shown that this configuration fulfills Bragg's law.
Three dimensions
Lattice with a basis
To compute structure factors for a specific lattice, compute the sum above over the atoms in the unit cell. Since crystals are often described in terms of their Miller indices, it is useful to examine a specific structure factor in terms of these.
Body-centered cubic (BCC)
As a convention, the body-centered cubic system is described in terms of a simple cubic lattice with primitive vectors  , with a basis consisting of
, with a basis consisting of  and
and  . The corresponding reciprocal lattice is also simple cubic with side .
. The corresponding reciprocal lattice is also simple cubic with side .
In a monatomic crystal, all the form factors are the same. The intensity of a diffracted beam scattered with a vector 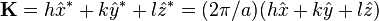 by a crystal plane with Miller indices
by a crystal plane with Miller indices  is then given by:
is then given by:
![{\begin{matrix}F_{{{\mathbf {K}}}}&=&f\left[e^{{-i{\mathbf {K}}\cdot {\vec {0}}}}+e^{{-i{\mathbf {K}}\cdot (a/2)({\hat {x}}+{\hat {y}}+{\hat {z}})}}\right]\\&=&f\left[1+e^{{-i{\mathbf {K}}\cdot (a/2)({\hat {x}}+{\hat {y}}+{\hat {z}})}}\right]\\&=&f\left[1+e^{{-i\pi (h+k+l)}}\right]\\&=&f\left[1+(-1)^{{h+k+l}}\right]\\\end{matrix}}](/2014-wikipedia_en_all_02_2014/I/media/5/1/f/f/51ffe8627d309a12bc0d33799fa66d47.png)
We then arrive at the following result for the structure factor for scattering from a plane :
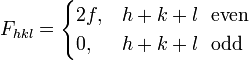
This result tells us that for a reflection to appear in a diffraction experiment involving a body-centered crystal, the sum of the Miller indices of the scattering plane must be even. If the sum of the Miller indices is odd, the intensity of the diffracted beam is reduced to zero due to destructive interference. This zero intensity for a group of diffracted beams is called a systematic absence. Since atomic form factors fall off with increasing diffraction angle corresponding to higher Miller indices, the most intense diffraction peak from a material with a BCC structure is typically the (110). The (110) plane is the most densely packed of BCC crystal structures and is therefore the lowest energy surface for a thin film to grow. Films of BCC materials like iron and tungsten therefore grow in a characteristic (110) orientation.
Face-centered cubic (FCC)
In the case of a monatomic FCC crystal, the atoms in the basis are at the origin with indices (0,0,0) and at the three face centers  ,
, 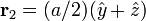 ,
,  with indices given by (1/2,1/2,0), (0,1/2,1/2), (1/2,0,1/2). An argument similar to the one above gives the expression
with indices given by (1/2,1/2,0), (0,1/2,1/2), (1/2,0,1/2). An argument similar to the one above gives the expression
![{\begin{matrix}F_{{{\mathbf {K}}}}&=&f\left[e^{{-i{\mathbf {K}}\cdot {\vec {0}}}}+e^{{-i{\mathbf {K}}\cdot (a/2)({\hat {x}}+{\hat {y}})}}+e^{{-i{\mathbf {K}}\cdot (a/2)({\hat {y}}+{\hat {z}})}}+e^{{-i{\mathbf {K}}\cdot (a/2)({\hat {x}}+{\hat {z}})}}\right]\\&=&f\left[1+(-1)^{{h+k}}+(-1)^{{k+l}}+(-1)^{{h+l}}\right]\\\end{matrix}}](/2014-wikipedia_en_all_02_2014/I/media/d/5/d/8/d5d86d80471822bbcc0ad76914229209.png)
with the result
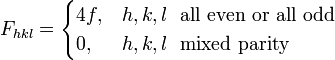
The most intense diffraction peak from a material that crystallizes in the FCC structure is typically the (111). Films of FCC materials like gold tend to grow in a (111) orientation with a triangular surface symmetry.
Diamond Crystal Structure
The Diamond cubic crystal structure occurs in diamond (carbon), most semiconductors and tin. The basis cell contains 8 atoms located at cell positions:


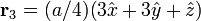
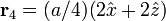
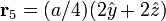
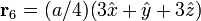
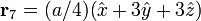
The Structure factor then takes on a form like this:
![{\begin{matrix}F_{{{\mathbf {K}}}}&=&f\left[{\begin{matrix}e^{{-i{\mathbf {K}}\cdot {\vec {0}}}}+e^{{-i{\mathbf {K}}\cdot (a/2)({\hat {x}}+{\hat {y}})}}+e^{{-i{\mathbf {K}}\cdot (a/2)({\hat {y}}+{\hat {z}})}}+e^{{-i{\mathbf {K}}\cdot (a/2)({\hat {x}}+{\hat {z}})}}+\\e^{{-i{\mathbf {K}}\cdot (a/4)({\hat {x}}+{\hat {y}}+{\hat {z}})}}+e^{{-i{\mathbf {K}}\cdot (a/4)(3{\hat {x}}+{\hat {y}}+3{\hat {z}})}}+e^{{-i{\mathbf {K}}\cdot (a/4)(3{\hat {x}}+3{\hat {y}}+{\hat {z}})}}+e^{{-i{\mathbf {K}}\cdot (a/4)({\hat {x}}+3{\hat {y}}+3{\hat {z}})}}\end{matrix}}\right]\\&=&f\left[{\begin{matrix}1+(-1)^{{h+k}}+(-1)^{{k+l}}+(-1)^{{h+l}}+\\(-i)^{{h+k+l}}+(-i)^{{3h+k+3l}}+(-i)^{{3h+3k+l}}+(-i)^{{h+3k+3l}}\end{matrix}}\right]\\&=&f\left[1+(-1)^{{h+k}}+(-1)^{{k+l}}+(-1)^{{h+l}}\right]\cdot \left[1+(-i)^{{h+k+l}}\right]\\\end{matrix}}](/2014-wikipedia_en_all_02_2014/I/media/5/e/9/3/5e939638ec24620062b34908c5745bf2.png)
with the result
- for mixed values (odds and even values combined) of h, k, and l, F2 will be 0
- if the values are unmixed and...
- h+k+l is odd then F=4f(1+i) or 4f(1-i), FF*=32f2
- h+k+l is even and exactly divisible by 4 (satisfies h+k+l=4n) then F = 8f
- h+k+l is even but not exactly divisible by 4(doesn't satisfy h+k+l=4n) then F = 0
Imperfect crystals
Although the perfect lattice is an extremely useful model, real crystals always exhibit imperfections, which can have profound effects on the structure and properties of the material. André Guinier [1] proposed a widely employed distinction between imperfections that preserve the long-range order of the crystal (disorder of the first kind) and those that destroy it (disorder of the second kind).
Disorder of the first kind
Disorder of the second kind
Liquids
In contrast with crystals, liquids have no long-range order (in particular, there is no regular lattice), so the structure factor does not exhibit sharp peaks. They do however show a certain degree of short-range order, depending on their density and on the strength of the interaction between particles. Liquids are isotropic, so that, after the averaging operation in Equation (4), the structure factor only depends on the absolute magnitude of the scattering vector 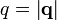 . For further evaluation, it is convenient to separate the diagonal terms
. For further evaluation, it is convenient to separate the diagonal terms 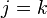 in the double sum, whose phase is identically zero, and therefore each contribute a unit constant:
in the double sum, whose phase is identically zero, and therefore each contribute a unit constant:
-
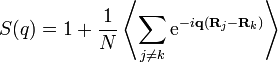 .
.(5)
One can obtain an alternative expression for in terms of the radial distribution function 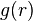 :[2]
:[2]
-
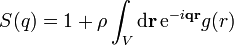 .
.(6)
Ideal gas
In the limiting case of no interaction, the system is an ideal gas and the structure factor is completely featureless:  , because there is no correlation between the positions
, because there is no correlation between the positions 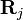 and
and 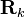 of different particles (they are independent random variables), so the off-diagonal terms in Equation (5) average to zero:
of different particles (they are independent random variables), so the off-diagonal terms in Equation (5) average to zero: ![\langle \exp[-i{\mathbf {q}}({\mathbf {R}}_{j}-{\mathbf {R}}_{k})]\rangle =\langle \exp(-i{\mathbf {q}}{\mathbf {R}}_{j})\rangle \langle \exp(i{\mathbf {q}}{\mathbf {R}}_{k})\rangle =0](/2014-wikipedia_en_all_02_2014/I/media/5/d/1/e/5d1e6dd89d0fbcea0a7dec0b31621659.png) .
.
High- limit
limit
Even for interacting particles, at high scattering vector the structure factor goes to 1. This result follows from Equation (6), since  is the Fourier transform of the "regular" function and thus goes to zero for high values of the argument . This reasoning does not hold for a perfect crystal, where the distribution function exhibits infinitely sharp peaks.
is the Fourier transform of the "regular" function and thus goes to zero for high values of the argument . This reasoning does not hold for a perfect crystal, where the distribution function exhibits infinitely sharp peaks.
Low- limit
In the low- limit, as the system is probed over large length scales, the structure factor contains thermodynamic information, being related to the isothermal compressibility 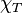 of the liquid by the compressibility equation:
of the liquid by the compressibility equation:
-
 .
.
Hard-sphere liquids

 from 1% to 40%.
from 1% to 40%.In the hard sphere model, the particles are described as impenetrable spheres with radius  ; thus, their center-to-center distance
; thus, their center-to-center distance  and they experience no interaction beyond this distance. Their interaction potential can be written as:
and they experience no interaction beyond this distance. Their interaction potential can be written as:
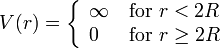
This model has an analytical solution[3] in the Percus–Yevick approximation. Although highly simplified, it provides a good description for systems ranging from liquid metals[4] to colloidal suspensions.[5] In an illustration, the structure factor for a hard-sphere fluid is shown in the Figure, for volume fractions from 1% to 40%.
Polymers
In polymer systems, the general definition (4) holds; the elementary constituents are now the monomers making up the chains. However, the structure factor being a measure of the correlation between particle positions, one can reasonably expect that this correlation will be different for monomers belonging to the same chain or to different chains.
Let us assume that the volume contains  identical molecules, each composed of
identical molecules, each composed of 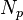 monomers, such that
monomers, such that 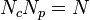 ( is also known as the degree of polymerization). We can rewrite (4) as:
( is also known as the degree of polymerization). We can rewrite (4) as:
-
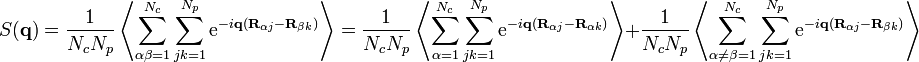 ,
,(7)
where indices 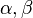 label the different molecules and
label the different molecules and  the different monomers along each molecule. On the right-hand side we separated intramolecular (
the different monomers along each molecule. On the right-hand side we separated intramolecular ( ) and intermolecular (
) and intermolecular (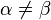 ) terms. Using the equivalence of the chains, (7) can be simplified:[6]
) terms. Using the equivalence of the chains, (7) can be simplified:[6]
-
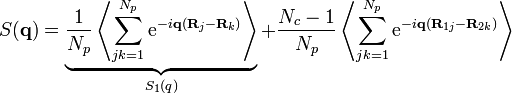 ,
,(8)
where 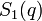 is the single-chain structure factor.
is the single-chain structure factor.
See also
Notes
- ↑ See Guinier, chapters 6-9
- ↑ See Chandler, section 7.5.
- ↑ Wertheim, M. (1963). "Exact Solution of the Percus-Yevick Integral Equation for Hard Spheres". Physical Review Letters 10 (8): 321. Bibcode:1963PhRvL..10..321W. doi:10.1103/PhysRevLett.10.321.
- ↑ Ashcroft, N.; Lekner, J. (1966). "Structure and Resistivity of Liquid Metals". Physical Review 145: 83. Bibcode:1966PhRv..145...83A. doi:10.1103/PhysRev.145.83.
- ↑ Pusey, P. N.; Van Megen, W. (1986). "Phase behaviour of concentrated suspensions of nearly hard colloidal spheres". Nature 320 (6060): 340. doi:10.1038/320340a0.
- ↑ See Teraoka, Section 2.4.4.
References
- Als-Nielsen, N. and McMorrow, D. (2011). Elements of Modern X-ray Physics (2nd edition). John Wiley & Sons.
- Guinier, A. (1963). X-ray Diffraction. In Crystals, Imperfect Crystals, and Amorphous Bodies. W. H. Freeman and Co.
- Chandler, D. (1987). Introduction to Modern Statistical Mechanics. Oxford University Press.
- Hansen, J. P. and McDonald, I. R. (2005). Theory of Simple Liquids (3rd edition). Academic Press.
- Teraoka, I. (2002). Polymer Solutions: An Introduction to Physical Properties. John Wiley & Sons.
External links
- Structure Factor Tutorial located at the University of York.