Quantitative genetics
| Part of a series on |
| Genetics |
|---|
| Key components |
| History and topics |
| Research |
|
|
| Biology portal |
Quantitative genetics is the study of the inheritance of continuously measured traits (such as height or weight) and their mechanisms. It can be an extension of simple Mendelian inheritance in that the combined effects of one or more genes and the environments in which they are expressed give rise to continuous distributions of phenotypic values.
Introduction
Quantitative genetics is that branch of population genetics which deals with phenotypes which vary continuously, and are best dealt with using Statistics (Biometrics). The phenotypes and underlying genotypes are summarised by measures of central tendency (e.g. the mean) and variability (e.g. the variance). Unlike other foci of Population Genetics, the phenotypes are not readily classifiable into "classes", of which a census may be ascertained. Some attributes may be analysable either way, depending upon the metric used to quantify them. Mendel himself had to discuss this matter in his famous paper, especially with respect to his attribute "length of stem".[1] Quantitative genetic analyses are heavily dependant on appropriate population Mating Systems, allowing zygotic frequencies to be specified appropriately from the gametic frequencies giving rise to them. Consequently, it can analyse phenotypic results arising from many situations, such as small mating-groups, gene-"islands", genetic drift, and inbreeding. It can analyse and "explain" heredity in its passage through both space (populations) and time (generations). It allows an understanding of methods used in plant and animal breeding, and of micro-evolution (e.g. natural selection).
History
The field was founded by the originators of the modern synthesis, R.A. Fisher, Sewall Wright and J. B. S. Haldane, and aimed to predict the response to selection given data on the phenotype and relationships of individuals.
Analysis of Quantitative Trait Loci, or QTL, is a more recent addition to the study of quantitative genetics. A QTL is a region in the genome that affects the trait or traits of interest. Quantitative trait loci approaches require accurate phenotypic, pedigree, and genotypic data from a large number of individuals.
Basic principles
The phenotypic value (P) of an individual is the combined effect of the genotypic value (G) and the environmental deviation (E):
- P = G + E
The genotypic value is the combined effect of all the genetic effects, including nuclear genes, mitochondrial genes, and interactions between the genes. It is worthwhile to note that the mathematics is related to the genetics: for which the brief following overview may be useful. In diploid organisms, a nucleus gene is represented twice in the genotype, one provided by each parent during sexual reproduction. Each gene is located at a particular place (a locus) on homologous chromosomes, one from each parent. Functional forms are called alleles. If both alleles at a gene have the same phenotypic effect, the gene is homozygous: if each allele at a gene is different, the gene is heterozygous. The average phenotypic outcome may be affected by dominance and by how genes interact with genes at other loci ("epistasis"). The founder of quantitative genetics - Sir Ronald Fisher - perceived all of this when he proposed the first mathematics of this branch of genetics.[2] He sought to define a single statistical summary of all the variance arising from phenotypic change during the course of genetic assortment and segregation, which he called "genetic" variance. His residual genotypic variance (called simply "residual" by Fisher) represented assortment which did not lead to phenotypic change. Subsequently, these partitions became known respectively as the "additive"(σ²A) and "dominance" (σ²D) variances, which titles do not appear to convey Fisher's partitions. However, the assortment/substitution partition of Fisher can be viewed as the average effects of genes after inheritance from parents (remember they pass through meiosis and fertilisation to be inherited - the very mechanisms effecting assortment/substitution).[3] So, "additive" actually means "average inherited effect", and is equivalent to Fisher's assortative disequilibrium. Recently it has been shown to contain the homozygote variance, a portion of the heterozygote variance, and a covariance between homozygote and heterozygote effects.[4] Falconer and Mackay next show that Fisher's "residual" (depicted in their Fig.7.2, p. 117) is due to heterozygosis, ie "dominance"; but not all of it, as some is embedded in the "additive" component, as we noted earlier.
This is not the only approach to defining and partitioning genotypic variances. An alternative was advanced by Mather and Jinks.[5] Their notation was entirely different to that of Fisher, and his method predominates. However, when their approach is translated into Fisher's notation, the relationships between the two approaches are clear. The Mather and Jinks approach is more "genetical" than Fisher's, being based on variances arising straightforwardly from homozygotes and heterozygotes. The inter-conversion between the Fisher and Mather & Jinks methods are given in Gordon (2003).
The mating-system assumed in deriving these genotypic variances is panmixia : which implies random fertilisation with uniform distribution of gametes in a very large population (theoretically, infinity). This rarely occurs in nature, as gamete distribution may be limited, for example by dispersal restrictions, or preferential matings, or chance sampling in small populations of gametes (gamodemes). Each gamete restriction leads to a descendant small-population (line). Individuals within a line will not all be the same, but they will be more similar than individuals from panmictic populations. In any source breeding group, many separate gamete restrictions will occur during a mating cycle, each one leading to a line. These lines also will vary with respect to their mean phenotypess, and the process is called dispersion. The inbreeding coefficient quantifies the increase in homozygosity which results. The values of this coefficient for a wide variety of situations (e.g. islands, "onion-skin" aggregates, linear strips, matings of related parents) are available.[6] As well as a general rise in homozygosity, the dispersed lines vary in their allele frequencies because of gamete sampling. However, the mean of the frequencies across all lines from the one source will be the same as the original frequencies in the source population. The phenotypic mean of all of these lines is less than that of the original source, this being inbreeding depression. The genetic variances also change relative to those of panmixia. Variance-within-lines decreases, but the variance-amongst-lines and the total-variance-in-the-system both increase (Mackay et al.; Gordon 2003). The first of these facts is common knowledge, but the latter two are not. Many of these lines will be inferior in phenotype: but, some lines will be superior, and some will be about average (Chapter 13 in Falconer et al.). Selection assisted by dispersion leads to maximum genetic advance (see previous references). Plant and animal breeders utilise these properties routinely, and have devised breeding methods especially to do so (e.g. line breeding, pure-line breeding, backcrossing).[7] The role of dispersion in natural selection has not received much attention.
The Environmental variance is much more straightforward. This can be subdivided into a pure environmental component (E) and an interaction component (I) describing the gene-environment interaction. The overall "single gene" model can be written as:
- P = a + d + E + I.
Expansion of the model to multiple genes (loci) is still not resolved satisfactorily, and until that is solved it is not possible to account for epistasis. The problem is being tackled currently. The contribution of those components cannot be determined in a single individual, but they can be estimated for whole populations by estimating the variances for those components, denoted as:
- VP = Va + Vd + VE + VI
The heritability of a trait is the proportion of the total (i.e. phenotypic) variance (VP) that is explained by the total genotypic variance (VG). This is known also as the "broad sense" heritability (H2). If only Additive genetic variance (VA) is used in the numerator, the heritability is "narrow sense" (h2). The broadsense heritability indicates the genotypic determination of the phenotype: while the latter estimates the degree of assortative disequlibrium in the trait. Fisher proposed that this narrow-sense heritability might be appropriate in considering the results of natural selection, focusing as it does on disequilibrium: and it has been used also for predicting the results of selection.
Resemblance between relatives
Central in estimating the variances for the various components is the principle of relatedness. A child has a father and a mother. Consequently, the child and father share 50% of their alleles, as do the child and the mother. However, the mother and father normally do not share alleles as a result of shared ancestors. Similarly, two full siblings share also on average 50% of the alleles with each other, while half siblings share only 25% of their alleles. This variation in relatedness can be used to estimate which proportion of the total phenotypic variance (VP) is explained by the above-mentioned components.
The principle of relationship (R) is central to understanding the resemblances within families and can be useful when calculating inbreeding. Relationship has two definitions that can be applied: -The probable portion of genes that are the same for two individuals due to common ancestry exceeding that of the base population -Additive/numerator relationship: the relationship coefficient (Rxy¬) = twice the probability of two genes at loci in different individuals being identical by descent. Rxy values can range from 0 to 2. Relationship can be calculated in several ways; from the known relationships of the individual, from bracket pedigrees, and from pedigree path diagrams.
Calculating relationship from known relationships
| Relationship | Relationship Coefficient |
|---|---|
| Individual and itself | 1.00 |
| Individual and a monozygotic twin | 1.00 |
| Individual and parent | 0.50 |
| Full siblings | 0.50 |
| Half siblings | 0.25 |
| Individual and grandparent | 0.25 |
| Son of sire and granddaughter of sire | 0.125 |
| Grandson and granddaughter of sire | 0.0625 |
- Note: if the common ancestor is inbred, multiply the relationship by (1+inbreeding coefficient)
Calculating relationship from pathway diagrams
RXY = Σ(.5)n(1+FCA)
n = number of segregations between X and Y through their common ancestor FCA = the inbreeding coefficient of the common ancestor
Example: calculating RAE and RBE Note: valid pathways only go through ancestors (only go against the direction of the arrow). For example, to calculate the relationship of A and B, the pathway A-D-B would be acceptable, whereas the pathway A-X-B would be not. The reason behind this is that having progeny together does not make two individuals related.
RAB: there are two possible pathways from A to E. A-D-F-E = (1/2)3 = .125 A-D-E = (1/2)2 = .25 Total: .375
RBE: there are four possible pathways from B to E. B-D-E = (1/2)2 = .25 B-D-F-E = (1/2)3 = .125 B-C-D-E = (1/2)3 = .125 B-C-D-F-E = (1/2)4 = .0625 Total: .5625
Heritability and repeatability
Heritability in the broad sense (H^2) measures the total genetic influence on phenotype. It takes into account additive genetics, dominance, and epistatic genetic effects. The more specific measure of heritability (h^2) is generally more useful in production because only additive effects are passed from one generation to the next, and h^2 represents the average proportion of differences due to additive genetics. The square root of h^2 equals the correlation between additive genotype and expressed phenotype. Low heritability leads to higher environmental influence, while high heritability can lead to more rapid genetic progress. Typically, heritability of reproductive traits is low, disease resistance and production are moderately low to moderate, and conformation is high.
Heritability can be calculated by taking the variance of additive genetics and dividing it by the variance of the phenotype.
h^2 = σ^2a/σ^2p
σ^2p is the sum of the variances of additive genetics, dominance, epistasis, and environmental effects, and can also be written as σ^2g.
Repeatability (r) is the average proportion of differences likely to be repeated in later records. This value can only be determined for traits that manifest multiple times in an animal’s lifetime, such as adult body mass, metabolic rate or litter size. Birth mass would not have a repeatability value. Generally, but not always, repeatability indicates the upper level of the heritability. Repeatability is calculated by taking the sum of genetic variance (σ^2g) and permanent environment variance, and dividing by the phenotypic variance.
r = (σ^2g + σ^2pe)/σ^2p
The above concept of repeatability is problematic for traits that necessarily change greatly between measurements. For example, body size increases greatly in most organisms between birth and adult-hood. Nonetheless, at a given age (or lifecycle stage), the relative size of individuals within a population could still be relatively consistent from time-to-time. For such traits, more complicated calculations can be used or pairs of measures can be compared with a Pearson product-moment correlation coefficient.
Correlated traits
Although some genes have only an effect on a single trait, many genes have an effect on various traits, which is termed pleiotropy. Because of this, a change in a single gene will have an effect on all those traits. This is calculated using covariances, and the phenotypic covariance (CovP) between two traits can be partitioned in the same way as the variances described above. The genetic correlation is calculated by dividing the covariance between the additive genetic effects of two traits by the square root of the product of the variances for the additive genetic effects of the two traits:
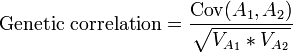
See also
References
- ↑ Mendel, Gregor (1865). "transl Experiments in plant hybridization". Verhandlungen naturforschender Verain iv.
- ↑ Fisher, R.A. (1930 (facsimile 1999)). The Genetic theory of natural selection. Oxford Clarendon Press. ISBN 0-19-850440-3.
- ↑ Falconer, et al (1996). Introduction to quantitative genetics. Longman. ISBN 0582-24302-5.
- ↑ Gordon, I.L. (2003). "Refinements to the partitioning of the inbred genotypic variance". Heredity 91 (1): 85–89. doi:10.1038/sj.hdy.6800284. PMID 12815457.
- ↑ Mather, et al (1971). Biometrical genetics. Chapman and Hall. ISBN 0-412-10220-X.
- ↑ Wright, Sewall (1951). "The genetical structure of populations". Annals of Eugenics 15: 323–354. doi:10.1111/j.1469-1809.1949.tb02451.x.
- ↑ Allard, R.W. (1960). Principles of plant breeding. Wiley. ISBN none Check
|isbn=value (help).
- Allard R.W. (1951). Principles of Plant Breeding. Wiley, New York. {a classic}
- Dohm, M.R. (2002). Repeatability estimates do not always set an upper limit to heritability. Functional Ecology 16: 273-280.
- Falconer, D. S. & Mackay TFC (1996). Introduction to Quantitative Genetics. Fourth edition. Addison Wesley Longman, Harlow, Essex, UK.
- Fisher R.A. (1930). The Genetical Theory of Natural Selection. Clarendon Press, Oxford, UK.
- Gordon I.L. (2003). Refinements to the partitioning of the inbred genotypic variance. Heredity 91: 85-89.
- Lynch M & Walsh B (1998). Genetics and Analysis of Quantitative Traits. Sinauer, Sunderland, MA.
- Mather K. & Jinks J.L. (1971). Biometrical Genetics. Chapman & Hall, London.
- Roff DA (1997). Evolutionary Quantitative Genetics. Chapman & Hall, New York.
- Seykora, Tony. Animal Science 3221 Animal Breeding. Tech. Minneapolis: University of Minnesota, 2011. Print.
- Wright S. (1951). The genetical structure of populations. Annals of Eugenics 15: 323-354.
External links
- Quantitative Genetics Resources by Michael Lynch and Bruce Walsh, including the two volumes of their textbook, Genetics and Analysis of Quantitative Traits and Evolution and Selection of Quantitative Traits.
- Resources by Nick Barton et al. from the textbook, Evolution.
| ||||||||
| |||||||||||||||||