Prader–Willi syndrome
| Prader–Willi Syndrome | |
|---|---|
| Classification and external resources | |
 People with Prader-Willi Syndrome, exhibiting characteristic facial appearance including narrow temples, an elongated face, thin upper lip, and a prominent nose. | |
| ICD-10 | Q87.1 |
| ICD-9 | 759.81 |
| OMIM | 176270 |
| DiseasesDB | 10481 |
| MedlinePlus | 001605 |
| eMedicine | ped/1880 |
| MeSH | D011218 |
| GeneReviews | |
Prader–Willi syndrome (/ˈprɑːdər ˈvɪli/; abbreviated P.W.S) is a rare genetic disorder in which seven genes (or some subset thereof) on chromosome 15 (q 11–13) are deleted or unexpressed (chromosome 15q partial deletion) on the paternal chromosome. It was first described in 1956 by Andrea Prader (1919–2001), Heinrich Willi (1900–1971), Alexis Labhart (1916–1994)), Andrew Ziegler, and Guido Fanconi of Switzerland.[2] Characteristic of PWS is "low muscle tone, short stature, incomplete sexual development, cognitive disabilities, problem behaviors, and a chronic feeling of hunger that can lead to excessive eating and life-threatening obesity."[3] The incidence of PWS is between 1 in 25,000 and 1 in 10,000 live births.
The maternal origin of the genetic material that is affected in the syndrome is important because the particular region of chromosome 15 involved is subject to parent of origin imprinting, meaning that for a number of genes in this region only one copy of the gene is expressed while the other is silenced through imprinting. For the genes affected in PWS, it is the maternal copy that is usually imprinted (and thus is silenced), while the mutated paternal copy is not functional. This means that while most people have one working and one silenced set of these genes, people with PWS have a non-working set and a silenced set. If the maternally derived genetic material from the same region is affected instead, the sister syndrome Angelman syndrome is the result.[4]
With the recent benefits of early diagnosis and ongoing interventions, the obesity rate among children with Prader–Willi Syndrome has decreased to be similar to the typical population. With behavioural therapy and other treatments, the effects of the syndrome can be reduced.[5]
Signs and symptoms
There are many signs and symptoms of Prader-Willi Syndrome. The variety of symptoms can range from poor muscle tone during infancy to behavioral problems in early childhood. Some symptoms that are usually found in infants, besides poor muscle tone, would be a lack of eye coordination, some are born with almond-shaped eyes, and due to poor muscle tone the infant may fail to have a strong sucking reflex. Their cry is weak, as well as the difficulty of waking up. another sign of this disease is a thin upper lip.
Holm et al. (1993) describe the following features and signs as pretest indicators of PWS, although not all will be present.
Uterus and birth
- Reduced fetal movement
- Frequent abnormal fetal position
- Occasional polyhydramnios (excessive amniotic fluid)
- Often breech or caesarean births
- Lethargy
- Hypotonia
- Feeding difficulties (due to poor muscle tone affecting sucking reflex)
- Difficulties establishing respiration
- Hypogonadism
Childhood
- Delayed milestones/intellectual delay
- Excessive sleeping
- Strabismus ('crossed eyes')
- Scoliosis (often not detected at birth)
- Cryptorchidism
- Speech delay
- Poor physical coordination
- Hyperphagia (over-eating) begins between the age of 2 and 8, and continues on throughout adulthood. Note change from feeding difficulties in infancy
- Excessive weight gain
- Sleep disorders
- Delayed puberty
- Short stature
- Obesity
- Extreme flexibility
Adulthood
- Infertility (males and females)
- Hypogonadism
- Sparse pubic hair
- Obesity
- Hypotonia (low muscle tone)
- Learning disabilities/borderline intellectual functioning (but some cases of average intelligence)
- Prone to diabetes mellitus
- Extreme flexibility
Physical appearance
- Prominent nasal bridge
- Small hands and feet with tapering of fingers
- Soft skin, which is easily bruised
- Excess fat, especially in the central portion of the body
- High, narrow forehead
- Thin upper lip
- Downturned mouth
- Almond-shaped eyes
- Light skin and hair relative to other family members
- Lack of complete sexual development
- Frequent skin picking
- Striae
- Delayed motor development
Neuro-cognitive
Individuals with PWS are at risk of learning and attention difficulties. Curfs and Fryns (1992) conducted research into the varying degrees of learning disability found in PWS.[6] Their results, using a measure of IQ, were as follows:
- 5%: IQ above 85 (high to low average intelligence)
- 27%: IQ 70 – 85 (borderline intellectual functioning)
- 39%: IQ 50 – 70 (mild intellectual disability)
- 27%: IQ 35 – 50 (moderate intellectual disability)
- 1%: IQ 20 – 35 (severe intellectual disability)
- <1%: IQ <20 (profound intellectual disability)
Cassidy found that 40% of individuals with PWS have borderline/low average intelligence,[7] a figure higher than the 32% found in Curfs and Fryns' study.[6] However, both studies suggest that most individuals (50–65%) fall within the mild/borderline/low average intelligence range.
Children with PWS show an unusual cognitive profile. They are often strong in visual organization and perception, including reading and vocabulary, but their spoken language (sometimes affected by hypernasality) is generally poorer than their comprehension. A marked skill in completing jigsaw puzzles has been noted,[8][9] but this may be an effect of increased practice.[10]
Auditory information processing and sequential processing are relatively poor, as are arithmetic and writing skills, visual and auditory short-term memory and auditory attention span. These sometimes improve with age, but deficits in these areas remain throughout adulthood.[8]
Behavioral
Prader–Willi syndrome is also frequently associated with an extreme and insatiable appetite, often resulting in morbid obesity. It is the most common genetic cause of morbid obesity in children.[11] There is currently no consensus as to the cause for this particular symptom, although genetic abnormalities in chromosome 15 disrupt the normal functioning of the hypothalamus.[7] Given that the hypothalamus arcuate nucleus regulates many basic processes, including appetite, there may well be a link. In the hypothalamus of people with PWS, nerve cells that produce oxytocin, a hormone thought to contribute to satiety, have been found to be abnormal.
People with Prader–Willi syndrome have high ghrelin levels, which are thought to directly contribute to the increased appetite, hyperphagia, and obesity seen in this syndrome. Cassidy states the need for a clear delineation of behavioral expectations, the reinforcement of behavioural limits and the establishment of regular routines.[21]
The main mental health difficulties experienced by people with PWS include compulsive behaviour (usually manifested in skin-picking) and anxiety.[8][12] Psychiatric symptoms, for example, hallucinations, paranoia and depression, have been described in some cases[8] and affect approximately 5–10% of young adults.[7] Psychiatric and behavioural problems are the most common cause of hospitalization.[13]
Endocrine
There are several aspects of PWS that support the concept of growth hormone deficiency in individuals with PWS. Specifically, individuals with PWS have short stature, are obese with abnormal body composition, have reduced fat free mass (FFM), have reduced lean body mass (LBM) and total energy expenditure, and have decreased bone density.
PWS is characterized by hypogonadism. This is manifested as undescended testes in males and benign premature adrenarche in females. Testes may descend with time or can be managed with surgery or testosterone replacement. Adrenarche may be treated with hormone replacement therapy.
Ophthalmologic
PWS is commonly associated with development of strabismus. In one study,[14] over 50% of patients had strabismus, mainly esotropia.
Genetics

PWS is caused by the deletion of the paternal copies of the SNRPN and necdin genes along with clusters of snoRNAs: SNORD64, SNORD107, SNORD108 and two copies of SNORD109, 29 copies of SNORD116 (HBII-85) and 48 copies of SNORD115 (HBII-52). These are on chromosome 15 located in the region 15q11-13.[15][16][17] This so-called PWS/AS region may be lost by one of several genetic mechanisms which, in the majority of instances occurs through chance mutation. Other less common mechanisms include; uniparental disomy, sporadic mutations, chromosome translocations, and gene deletions. Due to imprinting, the maternally inherited copies of these genes are virtually silent, only the paternal copies of the genes are expressed. PWS results from the loss of paternal copies of this region. Deletion of the same region on the maternal chromosome causes Angelman syndrome (AS). PWS and AS represent the first reported instances of imprinting disorders in humans.
The risk to the sibling of an affected child of having PWS depends upon the genetic mechanism which caused the disorder. The risk to siblings is <1% if the affected child has a gene deletion or uniparental disomy, up to 50% if the affected child has a mutation of the imprinting control region, and up to 25% if a parental chromosomal translocation is present. Prenatal testing is possible for any of the known genetic mechanisms.
A microdeletion in one family of the snoRNA HBII-52 has excluded it from playing a major role in the disease.[18]
Studies of human and mouse model systems have shown that deletion of the 29 copies of the C/D box snoRNA SNORD116 (HBII-85) has been shown to be the primary cause of Prader–Willi syndrome.[19][20][21][22][23]
Diagnosis
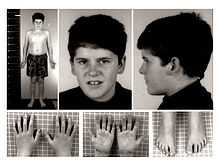
PWS affects approximately 1 in 10,000 to 1 in 25,000 newborns.[24] There are more than 400,000 people who live with PWS around the world.[25] It is traditionally characterized by hypotonia, short stature, hyperphagia, obesity, behavioral issues (specifically OCD-like behaviors), small hands and feet, hypogonadism, and mild mental retardation.[24] However, with early diagnosis and early treatment (such as with growth hormone therapy), the prognosis for persons with PWS is beginning to change. Like autism, PWS is a spectrum disorder and so symptoms can range from mild to severe, and may change throughout the person's lifetime. Various organ systems are affected.
Traditionally, Prader–Willi syndrome was diagnosed by clinical presentation. Currently, the syndrome is diagnosed through genetic testing; testing is recommended for newborns with pronounced hypotonia. Early diagnosis of PWS allows for early intervention as well as the early prescription of growth hormone. Daily recombinant growth hormone (GH) injections are indicated for children with PWS. GH supports linear growth and increased muscle mass, and may lessen food preoccupation and weight gain.
The mainstay of diagnosis is genetic testing, specifically DNA-based methylation testing to detect the absence of the paternally contributed Prader–Willi syndrome/Angelman syndrome (PWS/AS) region on chromosome 15q11-q13. Such testing detects over 97% of cases. Methylation-specific testing is important to confirm the diagnosis of PWS in all individuals, but especially those who are too young to manifest sufficient features to make the diagnosis on clinical grounds or in those individuals who have atypical findings.
Prader–Willi syndrome is often misdiagnosed as a variety of other syndromes due to many in the medical community's unfamiliarity with PWS.[11] Sometimes it is misdiagnosed as Down syndrome, simply because of the relative frequency of Down syndrome compared to PWS.[11]
Treatment
Prader–Willi syndrome has no cure; however, several treatments are in place to lessen the condition's symptoms. During infancy, subjects should undergo therapies to improve muscle tone. Speech and occupational therapy are also indicated. During the school years, children benefit from a highly structured learning environment as well as extra help. The largest problem associated with the syndrome is severe obesity.
Prescription of daily recombinant growth hormone injections are indicated for children with PWS. GH supports linear growth and increased muscle mass, and may lessen food preoccupation and weight gain.[26][27][28]
Because of severe obesity, obstructive sleep apnea is a common sequela, and a positive airway pressure machine is often needed. There may come a time when a person who has been diagnosed with PWS may have to undergo surgical procedures. One surgery in particular has proven to be unsuccessful for treating the obesity, is the gastric bypass. Patients with Prader–Willi syndrome have a very high tolerance to pain; therefore they may be experiencing significant abdominal symptoms such as acute gastritis, appendicitis, or cholecystitis and not be aware of it until later.
Society and culture
%2C_de_Juan_Carre%C3%B1o_de_Miranda..jpg)
Despite its rarity, Prader–Willi syndrome has been often referenced in popular culture, partly due to the fascination surrounding the insatiable appetite and obesity that are symptoms of the syndrome.
Prader–Willi syndrome has been depicted and documented several times in television. It appeared in the UK media in July 2007 when Channel 4 aired a program Can't Stop Eating, surrounding the everyday lives of two people with Prader–Willi syndrome, Joe and Tamara.[29] An individual with Prader-Willi Syndrome featured in the episode entitled "Dog Eat Dog" of the television series CSI: Crime Scene Investigation (aired on November 24, 2005).[30]
In December 2011 the Taipei Times, in Taiwan, highlighted the tragedy of a taxi driver who had killed himself and his nine-year-old daughter who had the condition, in what police described as a "probable murder-suicide."[31]
Actress Mayim Bialik—former star of TV's Blossom and currently playing a neuroscientist named Amy Farrah Fowler on The Big Bang Theory—completed her PhD in 2007 with a dissertation on Prader–Willi syndrome.[32][33]
See also
References
- ↑ Mary Jones. "Case Study: Cataplexy and SOREMPs Without Excessive Daytime Sleepiness in Prader Willi Syndrome. Is This the Beginning of Narcolepsy in a Five Year Old?". European Society of Sleep Technologists. Retrieved April 6, 2009.
- ↑ synd/1836 at Who Named It?
- ↑ "Questions and Answers on Prader-Willi Syndrome". Prader-Willi Syndrome Association. Retrieved February 2, 2012.
- ↑ http://www.nichd.nih.gov/health/topics/prader-willi/conditioninfo/Pages/faqs.aspx
- ↑ Butler, Merlin Gene (2006). Management of Prader-Willi Syndrome. Springer. ISBN 978-0-387-25397-8.
- ↑ 6.0 6.1 Curfs LM, Fryns JP (1992). "Prader-Willi syndrome: a review with special attention to the cognitive and behavioral profile". Birth Defects Orig. Artic. Ser. 28 (1): 99–104. PMID 1340242.
- ↑ 7.0 7.1 7.2 Cassidy SB (1997). "Prader-Willi syndrome". Journal of Medical Genetics 34 (11): 917–23. doi:10.1136/jmg.34.11.917. PMC 1051120. PMID 9391886.
- ↑ 8.0 8.1 8.2 8.3 Udwin O (November 1998). "Prader-Willi syndrome: Psychological and behavioural characteristics". Contact a Family.
- ↑ Holm VA, Cassidy SB, Butler MG, et al. (1993). "Prader-Willi syndrome: consensus diagnostic criteria". Pediatrics 91 (2): 398–402. PMID 8424017.
- ↑ Whittington J, Holland A, Webb T, Butler J, Clarke D, Boer H (February 2004). "Cognitive abilities and genotype in a population-based sample of people with Prader-Willi syndrome". J Intellect Disabil Res 48 (Pt 2): 172–87. doi:10.1111/j.1365-2788.2004.00556.x. PMID 14723659.
- ↑ 11.0 11.1 11.2 Nordqvist, Christian (15 Mar 2010). "What Is Prader-Willi Syndrome? What Causes Prader-Willi Syndrome?". Medical News Today. MediLexicon International. Retrieved 4 December 2012.
- ↑ Clark DJ, Boer H, Webb T (1995). "General and behavioural aspects of PWS: a review". Mental Health Research 8 (195): 38–49.
- ↑ Cassidy SB, Devi A, Mukaida C (1994). "Aging in PWS: 232 patients over age 30 years". Proc. Greenwood Genetic Centre 13: 102–3.
- ↑ Hered RW, Rogers S, Zang YF, Biglan AW (1988). "Ophthalmologic features of Prader-Willi syndrome". J Pediatr Ophthalmol Strabismus 25 (3): 145–50. PMID 3397859.
- ↑ Online 'Mendelian Inheritance in Man' (OMIM) Prader-Willi Syndrome; PWS -17627
- ↑ de los Santos T, Schweizer J, Rees CA, Francke U (November 2000). "Small evolutionarily conserved RNA, resembling C/D box small nucleolar RNA, is transcribed from PWCR1, a novel imprinted gene in the Prader-Willi deletion region, which Is highly expressed in brain". American Journal of Human Genetics 67 (5): 1067–82. doi:10.1086/303106. PMC 1288549. PMID 11007541.
- ↑ Cavaillé J, Buiting K, Kiefmann M, et al. (December 2000). "Identification of brain-specific and imprinted small nucleolar RNA genes exhibiting an unusual genomic organization". Proc. Natl. Acad. Sci. U.S.A.. 97 (26): 14311–6. doi:10.1073/pnas.250426397. PMC 18915. PMID 11106375.
- ↑ Runte M, Varon R, Horn D, Horsthemke B, Buiting K (2005). "Exclusion of the C/D box snoRNA gene cluster HBII-52 from a major role in Prader-Willi syndrome.". Hum Genet 116 (3): 228–30. doi:10.1007/s00439-004-1219-2. PMID 15565282.
- ↑ Skryabin BV, Gubar LV, Seeger B, et al. (2007). "Deletion of the MBII-85 snoRNA gene cluster in mice results in postnatal growth retardation". PLoS Genet. 3 (12): e235. doi:10.1371/journal.pgen.0030235. PMC 2323313. PMID 18166085.
- ↑ Sahoo T, del Gaudio D, German JR, Shinawi M, Peters SU, Person RE, Garnica A, Cheung SW, Beaudet AL (2008). "Prader-Willi phenotype caused by paternal deficiency for the HBII-85 C/D box small nucleolar RNA cluster.". Nat Genet 40 (6): 719–21. doi:10.1038/ng.158. PMC 2705197. PMID 18500341.
- ↑ Ding F, Li HH, Zhang S, Solomon NM, Camper SA, Cohen P, Francke U (2008). "SnoRNA Snord116 (Pwcr1/MBII-85) deletion causes growth deficiency and hyperphagia in mice". In Akbarian, Schahram. PLoS ONE 3 (3): e1709. doi:10.1371/journal.pone.0001709. PMC 2248623. PMID 18320030.
- ↑ Ding F, Prints Y, Dhar MS, Johnson DK, Garnacho-Montero C, Nicholls RD, Francke U (2005). "Lack of Pwcr1/MBII-85 snoRNA is critical for neonatal lethality in Prader-Willi syndrome mouse models". Mamm Genome 16 (6): 424–31. doi:10.1007/s00335-005-2460-2. PMID 16075369.
- ↑ de Smith AJ, Purmann C, Walters RG, et al. (June 2009). "A Deletion of the HBII-85 Class of Small Nucleolar RNAs (snoRNAs) is Associated with Hyperphagia, Obesity and Hypogonadism". Hum. Mol. Genet. 18 (17): 3257–65. doi:10.1093/hmg/ddp263. PMC 2722987. PMID 19498035.
- ↑ 24.0 24.1 Killeen, Anthony A. (2004). "Genetic Inheritance". Principles of Molecular Pathology. Humana Press. p. 41. ISBN 978-1-58829-085-4.
- ↑ [[http://www.aolhealth.com/bio/katherine-tweed|Tweed, Katherine]] (September 2009). "Shawn Cooper Struggles with Prader Willi Syndrome". AOL Health. Retrieved September 2009.
- ↑ Davies PS, Evans S, Broomhead S, et al. (May 1998). "Effect of growth hormone on height, weight, and body composition in Prader-Willi syndrome". Arch. Dis. Child. 78 (5): 474–6. doi:10.1136/adc.78.5.474. PMC 1717576. PMID 9659098.
- ↑ Carrel AL, Myers SE, Whitman BY, Allen DB (April 2002). "Benefits of long-term GH therapy in Prader-Willi syndrome: a 4-year study". J. Clin. Endocrinol. Metab. 87 (4): 1581–5. doi:10.1210/jc.87.4.1581. PMID 11932286.
- ↑ Höybye C, Hilding A, Jacobsson H, Thorén M (May 2003). "Growth hormone treatment improves body composition in adults with Prader-Willi syndrome". Clin. Endocrinol. (Oxf) 58 (5): 653–61. doi:10.1046/j.1365-2265.2003.01769.x. PMID 12699450.
- ↑ "Can't Stop Eating". 2006. Retrieved June 12, 2009.
- ↑ "Dog Eat Dog". www.csifiles.com. Retrieved June 12, 2009.
- ↑ Group urges more support for Prader-Willi sufferers - Taipei Times. Published Dec 24, 2011. Retrieved May 27, 2012.
- ↑ "Life After Child Stardom - Not by the Numbers". Abcnews.go.com. 2006-11-24. Retrieved 2012-05-27.
- ↑ Bialik, Mayim C. "Hypothalamic regulation in relation to maladaptive, obsessive-compulsive, affiliative, and satiety behaviors in Prader-Willi syndrome" (PhD Diss., UCLA, 2007).
External links
| Wikimedia Commons has media related to Prader-Willi syndrome. |
| |||||||||||||||||||