Position weight matrix
A position weight matrix (PWM), also known as a position-specific weight matrix (PSWM) or position-specific scoring matrix (PSSM), is a commonly used representation of motifs (patterns) in biological sequences.
PWMs are often derived from a set of aligned sequences that are thought to be functionally related and have become an important part of many software tools for computational motif discovery.
Background
The position weight matrix was introduced by American geneticist Gary Stormo and colleagues in 1982[1] as an alternative to consensus sequences. Consensus sequences had previously been used to represent patterns in biological sequences, but had difficulties in the prediction of new occurrences of these patterns.[2] The first use of PWMs was in the discovery of RNA sites that function as translation initiation sites. The perceptron algorithm was suggested by Polish American mathematician Andrzej Ehrenfeucht in order to create a matrix of weights which could distinguish true binding sites from other non-functional sites with similar sequences. Training the perceptron on both sets of sites resulted in a matrix and a threshold to distinguish between the two sets.[1] Using the matrix to scan new sequences not included in the training set showed that this method was both more sensitive and precise than the best consensus sequence.[2]
The advantages of PWMs over consensus sequences have made PWMs a popular method for representing patterns in biological sequences and an essential component in modern algorithms for motif discovery.[3][4]
Basic PWM with frequencies
A PWM has one row for each symbol of the alphabet: 4 rows for nucleotides in DNA sequences or 20 rows for amino acids in protein sequences. It also has one column for each position in the pattern. A basic PWM using relative frequencies is constructed by counting the occurrences of each symbol at each position and then normalising at each position. Formally, given a set X of N aligned sequences of length l, the elements of the PWM M are calculated:
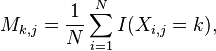
where i  (1,...,N), j (1,...,l), k is the set of symbols in the alphabet and I(a=k) is an indicator function where I(a=k) is 1 if a=k and 0 otherwise.
(1,...,N), j (1,...,l), k is the set of symbols in the alphabet and I(a=k) is an indicator function where I(a=k) is 1 if a=k and 0 otherwise.
For example, given the following DNA sequences:
GAGGTAAAC
TCCGTAAGT
CAGGTTGGA
ACAGTCAGT
TAGGTCATT
TAGGTACTG
ATGGTAACT
CAGGTATAC
TGTGTGAGT
AAGGTAAGT
the PWM using relative frequencies is:
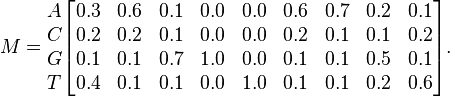
PWMs assume statistical independence between positions in the pattern, as the probabilities for each position are calculated independently of other positions. From the definition above, it follows that the sum of values for a particular position (that is, summing over all symbols) is 1. Each column can therefore be regarded as an independent multinomial distribution. This makes it easy to calculate the probability of a sequence given a PWM, by multiplying the relevant probabilities at each position. For example, the probability of the sequence S = GAGGTAAAC given the above PWM M can be calculated:
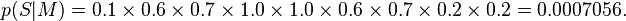
Pseudocounts (or Laplace estimators) are often applied to PWMs when a small dataset is used, in order to avoid matrix entries having a value of 0.[6] This is equivalent to multiplying each column of the PWM by a Dirichlet distribution and allows the probability to be calculated for new sequences (that is, sequences which were not part of the original dataset): in the example above, without pseudocounts, any sequence which did not have a G in the 4th position or a T in the 5th position would have a probability of 0, regardless of the other positions.
Using log likelihoods
Often the element in PWMs are calculated as log likelihoods. That is, the elements of the PWM are transformed using a background model  so that:
so that:
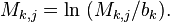
The simplest background model assumes that each letter appears equally frequently in the dataset. That is, the value of 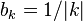 for all symbols in the alphabet (0.25 for nucleotides and 0.05 for amino acids). Applying this transformation to the PWM M from above gives:
for all symbols in the alphabet (0.25 for nucleotides and 0.05 for amino acids). Applying this transformation to the PWM M from above gives:

The  entries in the matrix make clear the advantage of adding pseudocounts, especially when using small datasets to construct M. The background model need not have equal values for each symbol: for example, when studying organisms with a high GC-content, the values for C and G may be increased with a corresponding decrease for the A and T values.
entries in the matrix make clear the advantage of adding pseudocounts, especially when using small datasets to construct M. The background model need not have equal values for each symbol: for example, when studying organisms with a high GC-content, the values for C and G may be increased with a corresponding decrease for the A and T values.
When the PWM elements are calculated using log likelihoods, the score of a sequence given a particular PWM can be calculated be adding (rather than multiplying) the relevant values at each position in the PWM. The sequence score gives an indication of how different the sequence is from a random sequence. The score is 0 if the sequence has the same probability of being a functional site and of being a random site. The score is greater than 0 if it is more likely to be a functional site than a random site, and less than 0 if it is more likely to be a random site than a functional site.[5] The sequence score can also be interpreted in a physical framework as the binding energy for that sequence.
Information content of a PWM
The information content (IC) of a PWM is sometimes of interest, as it says something about how different a given PWM is from a uniform distribution.
The self-information of observing a particular symbol at a particular position of the motif is:
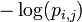
The expected (average) self-information of a particular element in the PWM is then:
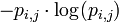
Finally, the IC of the PWM is then the sum of the expected self-information of every element:
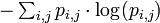
Often, it is more useful to calculate the information content with the background letter frequencies of the sequences you are studying rather than assuming equal probabilities of each letter (e.g., the GC-content of DNA of thermophilic bacteria range from 65.3 to 70.8,[7] thus a motif of ATAT would contain much more information than a motif of CCGG). The equation for information content thus becomes
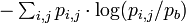
where  is the background frequency for that letter. This corresponds to the Kullback-Leibler divergence or relative entropy. However, it has been shown that when using PSSM to search genomic sequences (see below) this uniform correction can lead to overestimation of the importance of the different bases in a motif, due to the uneven distribution of n-mers in real genomes, leading to a significantly larger number of false positives.[8]
is the background frequency for that letter. This corresponds to the Kullback-Leibler divergence or relative entropy. However, it has been shown that when using PSSM to search genomic sequences (see below) this uniform correction can lead to overestimation of the importance of the different bases in a motif, due to the uneven distribution of n-mers in real genomes, leading to a significantly larger number of false positives.[8]
Using PWMs
There are various algorithms to scan for hits of PWMs in sequences. One example is the MATCH algorithm[9] which has been implemented in the ModuleMaster.[10] More sophisticated algorithms for fast database searching with nucleotide as well as amino acid PWMs/PSSMs are implemented in the possumsearch software and are described by Beckstette, et al. (2006).[11]
References
- ↑ 1.0 1.1 Stormo, Gary D.; Schneider, Thomas D.; Gold, Larry; Ehrenfeucht, Andrzej (1982). "Use of the ‘Perceptron’ algorithm to distinguish translational initiation sites in E. coli". Nucleic Acids Research 10 (9): 2997–3011. doi:10.1093/nar/10.9.2997.
- ↑ 2.0 2.1 Stormo, G. D. (1 January 2000). "DNA binding sites: representation and discovery". Bioinformatics 16 (1): 16–23. doi:10.1093/bioinformatics/16.1.16.
- ↑ Sinha, S. (27 July 2006). "On counting position weight matrix matches in a sequence, with application to discriminative motif finding". Bioinformatics 22 (14): e454–e463. doi:10.1093/bioinformatics/btl227.
- ↑ Xia, Xuhua (2012). "Position Weight Matrix, Gibbs Sampler, and the Associated Significance Tests in Motif Characterization and Prediction". Scientifica 2012: 1–15. doi:10.6064/2012/917540.
- ↑ 5.0 5.1 Guigo, Roderic. "An Introduction to Position Specific Scoring Matrices". http://bioinformatica.upf.edu. Retrieved 12 November 2013.
- ↑ Nishida, K.; Frith, M. C.; Nakai, K. (23 December 2008). "Pseudocounts for transcription factor binding sites". Nucleic Acids Research 37 (3): 939–944. doi:10.1093/nar/gkn1019.
- ↑ Aleksandrushkina NI, Egorova LA (1978). "Nucleotide makeup of the DNA of thermophilic bacteria of the genus Thermus". Mikrobiologiia 47 (2): 250–2. PMID 661633.
- ↑ Erill I, O'Neill MC (2009). "A reexamination of information theory-based methods for DNA-binding site identification". BMC Bioinformatics 10: 57. doi:10.1186/1471-2105-10-57. PMC 2680408. PMID 19210776.
- ↑ Kel AE, et al. (2003). "MATCHTM: a tool for searching transcription factor binding sites in DNA sequences". Nucleic Acids Research 31 (13): 3576–3579. doi:10.1093/nar/gkg585. PMC 169193. PMID 12824369.
- ↑ Wrzodek, Clemens; Schröder, Adrian; Dräger, Andreas; Wanke, Dierk; Berendzen, Kenneth W.; Kronfeld, Marcel; Harter, Klaus; Zell, Andreas (9 October 2009). "ModuleMaster: A new tool to decipher transcriptional regulatory networks". Biosystems (Ireland: Elsevier) 99 (1): 79–81. doi:10.1016/j.biosystems.2009.09.005. ISSN 0303-2647. PMID 19819296.
- ↑ Beckstette, M. et al. (2006). "Fast index based algorithms and software for matching position specific scoring matrices". BMC Bioinformatics 7: 389. doi:10.1186/1471-2105-7-389. PMC 1635428. PMID 16930469.
External links
- 3PFDB — a database of Best Representative PSSM Profiles (BRPs) of Protein Families generated using a novel data mining approach.
- UGENE — PSS matrices design, integrated interface to JASPAR, Uniprobe and SITECON databases.