Oxaziridine
| Oxaziridine | ||
|---|---|---|
 | ||
| IUPAC name Oxaziridine | ||
| Systematic name Oxaziridine[1] | ||
| Identifiers | ||
| PubChem | 15817734 | |
| ChemSpider | 10614170 | |
| Jmol-3D images | Image 1 | |
| ||
| ||
| Properties | ||
| Molecular formula | CH3NO | |
| Molar mass | 45.04 g mol−1 | |
| Except where noted otherwise, data are given for materials in their standard state (at 25 °C (77 °F), 100 kPa) | ||
| Infobox references | ||

An oxaziridine is an organic molecule that features a three-membered heterocycle containing oxygen, nitrogen, and carbon. Oxaziridne derivatives are commonly used as reagents in organic chemistry for a variety of oxidations, including alpha hydroxylation of enolates, epoxidation and aziridination of olefins, and other heteroatom transfer reactions. Oxaziridines are also synthetically useful as intermediates that undergo rearrangement to amides, as well as participate in [3+2] cycloadditions with various heterocumulenes to form substituted five remembered heterocycles. Chiral oxaziridine derivatives have also been developed in order to effect asymmetric oxygen transfer to prochiral enolates, as well as other substrates. Some oxaziridines also have the interesting property of a high barrier to inversion of the nitrogen, allowing for the possibility of chirality at the nitrogen atom.
History
Oxaziridine derivatives were first synthesized in the mid-1950s by Emmons[2] and subsequently by Krimm[3] and Horner and Jürgens.[4] Whereas oxygen and nitrogen typically act as nucleophiles due to their high electronegativity, oxaziridines allow for electrophilic transfer of both heteroatoms. This unusual reactivity is due to the presence of the highly strained three membered ring and the relatively weak N-O bond. Nucleophiles tend to attack at the aziridine nitrogen when the nitrogen substituent is small (R1= H), and at the oxygen atom when the nitrogen substituent has greater steric bulk. The unusual electronics of the oxaziridine system may be exploited to perform a number of oxygen and nitrogen transfer reactions including, but not limited to: α-hydroxylation of enolates, epoxidation of alkenes, selective oxidation of sulfides and selenides, amination of N-nucleophiles and N-acylamidation.
Chiral oxaziridine reagents have been developed, which allow for stereospecific transfer of heteroatoms. Chirality in oxaziridine compounds may be derived from the structure of the substituents on the oxaziridine, or from the conformationally locked nitrogen atom. Oxaziridines are unique in their exceptionally high inversion barrier for nitrogen to retain its stereochemical configuration. Chiral camphorsulfonyloxaziridines were synthesized by F. A. Davis in the 1970s have become a cornerstone of asymmetric synthesis. Among many prominent total syntheses employing oxaziridines, both the Holton Taxol total synthesis and the Wender Taxol total synthesis feature asymmetric α-hydroxylation with camphorsulfonyloxaziridine as a key step in the synthesis of Taxol, a complicated natural product marketed as a chemotherapy agent.
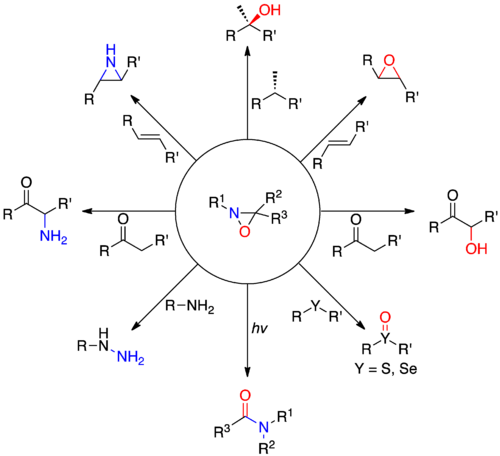
Synthesis
N-H, N-Alkyl, N-Aryloxaziridines
The two main approaches to synthesis of N-H, N-Alkyl, and N-Aryloxaziridines are oxidation of imines with peracids (A) and amination of carbonyls (B).

Additionally, oxidation of chiral imines and oxidation of imines with chiral peracids may yield enantiopure oxaziridines. Some oxaziridines have the unique property of configurationally stable nitrogen atoms at room temperature due to an inversion barrier of 24 to 31 kcal/mol. Enantiopure oxaziridines where stereochemistry is entirely due to configurationally stable nitrogen are reported.[5]
N-Sulfonyloxaziridines
In the late 1970s and early 1980s Franklin A. Davis synthesized the first N-sulfonyloxaziridines, which act exclusively as oxygen transfer reagents, and are the most predominantly used class of oxaziridines today.[6] While originally synthesized with mCPBA and the phase transfer catalyst benzyltrimethylammonium chloride, an improved synthesis using oxone as the oxidant is now most prevalent.[7]
There are many N-sulfonyloxaziridines used today, each with slightly different properties and reactivity. A summary of some of these reagents may be found in the table below.[8][9][10][11][12][13][14][15][16]

Perfluorinated oxaziridines
Perfluorinated oxaziridines display an interesting reactivity scheme distinct from hydrocarbon based oxaziridines. The highly electron withdrawing perfluoroalkyl substituents cause oxaziridines of this class to have reactivity more similar to dioxiranes than typical oxaziridines.[17] Notably, perfluoroalkyloxaziridines have the ability to hydroxylate certain C-H bonds with high selectivity. Perfluorinated oxaziridines may be synthesized by subjecting a perfluorinated imine to perfluoromethyl fluorocarbonyl peroxide and a metal fluoride to act as an HF scavenger.[17]

Reactions of oxaziridines
Oxygen transfer
α-Hydroxylation of enolates
α-hydroxyketones, or acyloins, are an important synthetic motif present in many natural products. α-hydroxyketones have been synthesized in many ways, including reduction of α-diketones, substitution of a hydroxyl for a leaving group and direct oxidation of an enolate. Oxodiperoxymolybdenum(pyridine)-(hexamethylphosphoric triamide) (MoOPH) and N-sulfonyloxaziridines are the most common electrophilic sources of oxygen implemented in this process. One advantage of using N-sulfonyloxaziridines is that higher chiral induction is almost invariably observed relative to MoOPH and other oxidants.[18] High yield (77-91%) and dr (95:5 - 99:1) are reported for α-hydroxylation with the Evans' chiral auxiliary with N-sulfonyloxaziridine as the electrophile.[18] Chiral induction has been demonstrated with many other chiral ketones and ketones with chiral auxiliaries, including SAMP and RAMP.[8]
{kind=link}

Davis has performed extensive work on asymmetric hydroxylation of prochiral enolates with camphorsulfonyloxaziridine derivatives, achieving moderate to high enantiomeric excess.[11] The commonly accepted proposed transition state that justifies this stereochemical outcome involves an open transition state where the steric bulk of R1 determines the face of approach.[8]

Interestingly, the selectivity of some hydroxylations may be drastically improved in some cases with the addition of coordinating groups alpha to the oxaziridine ring as oxaziridines 3b and 3c in the table above.[14] In these instances it is proposed that the reaction proceeds through a closed transition state where the metal oxyanion is stabilized by chelation from the sulfate and coordinating groups on the camphor skeleton.[8]

α-Hydroxylation with oxaziridines has been widely implemented in total synthesis. It is a key step in both the Holton Taxol total synthesis and the Wender Taxol total synthesis. Additionally, Forsyth implemented the transformation in his synthesis of the C3-C14 (Substituted 1,7-Dioxaspiro[5.5]undec-3-ene) System of Okadaic acid.[19]

Epoxidation of alkenes
Epoxidation of alkenes is a versatile synthetic technique in organic synthesis. Epoxides may be derivatized to a number of useful functional groups and motifs. Classically, epoxidation is carried out with mCPBA or other peracids. Oxaziridines have been found to perform similar chemistry, and are useful for the formation of highly acid sensitive epoxides.[5] Papeo et al. performed a synthesis of (-)-Chaetominine that utilized oxaziridine epoxidation as a late stage transformation as seen below.[20]

Another transformation of high synthetic utility is asymmetric epoxidation. There exist a number of asymmetric epoxidations in the literature, including the Sharpless epoxidation, the Jacobsen-Katsuki epoxidation and the Juliá-Colonna Epoxidation. These methods have one major drawback in that they require very specific functionality in order to achieve selectivity. The Sharpless epoxidation is specific to allylic alcohols, the Jacobsen epoxidation requires cis-disubstituted aryl alkenes, and the Juliá epoxidation requires α-β unsaturated ketones. Epoxidation with asymmetric oxaziridine reagents is among a select few transformations that are stereospecific to unfunctionalized alkenes with sufficient asymmetric induction to provide steric differentiation between faces.[5] It has even been shown to be possible to produce stereospecific epoxidation catalytic in the oxaziridine chiral unit. Further investigation into these reactions may be required before levels of enantiometic excess become practical for large scale synthesis. Lusinichi et al. have investigated asymmetric epoxidation with a chiral oxaziridinium salt using oxone as the stoichiometric oxidant seen below.[21]

Hydroxylation of unactivated hydrocarbons
Perfluorinated oxaziridines are known to hydroxylate unactivated hydrocarbons with remarkable regio, and diastereospecificity.[17] This is a highly coveted transformation, and similar reactivity and specificity is seldom rivaled, especially considering the nonmetallic nature of the oxidant. Perfluorinated oxaziridines show high selectivity toward tertiary hydrogens. Hydroxylation of primary carbons and dihydroxylation of a compound with two oxidizable sites have never been observed. Retention of stereochemistry is very high, often 95 - 98%. (retenton of stereochemistry may be further enhanced by the addition of a fluoride salt).[22]

Nitrogen transfer
Considerably less attention has been given to oxaziridines as nitrogen transfer reagents. Oxaziridines with unsubstituted or acylated nitrogens are capable of performing this chemistry. The first instance of nitrogen transfer was recorded in 1964 by Ohme and Schmitz.[23]
Amination of N-nucleophiles
Amination of nucleophiles with N-unsubstituted oxaziridines is quite versatile in the breadth of possible nucleophiles and corresponding products. Hydrazines may be derived from the amination of secondary or tertiary amines, hydroxylamine and thiohydroxamines may be formed from their corresponding alcohols and thiols, sulfimides may be formed from thioethers and α-aminoketones may be formed by attack of corresponding enolates.[24]

N-acylamidation
The transfer of acylated amines is more difficult than that of unsubstituted amines, although, unlike amine transfer by oxaziridines, there are no alternative methods that directly transfer acylated amines.[24] Acylamine transfer has primarily been performed using amines and hydrazines as nucleophiles. Very few transfers of acylated nitrogens to carbon nucleophiles have been successfully performed, although some do exist in the literature.[24]
Rearrangements
Oxaziridines have been found to undergo rearrangement reactions via a radical mechanism when irradiated with UV light or in the presence of a single electron transfer reagent such as CuI. spirocylic oxaziridines undergo ring expansions to the corresponding lactam.[25] Interestingly, the migrating substituent is determined by a stereoelectronic effect where the group trans to the lone pair on the nitrogen will always be the predominant migration product.[26] In light of this effect, it is possible to take advantage of the chiral nitrogen due to high inversion barrier to direct the rearrangement. This phenomenon is demonstrated by observed selectivities in the rearrangements below. In the rearrangement on the left the thermodynamically unfavorable product is observed exclusively, while in the reaction on the right the product derived from the less stable radical intermediate is favored.[25]

Aubé takes advantage of this rearrangement as the key step in his synthesis of (+)-yohimbine,[25] a natural medicine classified by the NIH as possibly effective in the treatment of erectile dysfunction and the sexual problems caused by selective serotonin reuptake inhibitors.[27]

It is also notable that oxaziridines will thermally rearrange to nitrones. Cis-trans selectivity of the resulting nitrone is poor, however, yields are good to excellent. It is thought that some oxaziridines racemize over time through a nitrone intermediate.[5]

Cycloaddions with heterocumulenes
Oxaziridines undergo cycloaddition reactions with heterocumulenes to afford a number of unique five membered heterocycles, as depicted in the figure below. This reactivity is due to the strained three membered ring and weak N-O bond.[5]

References
- ↑ "CID 15817734 - PubChem Public Chemical Database". The PubChem Project. USA: National Center for Biotechnology Information.
- ↑ Emmons, W. D. (1956). J. Am. Chem. Soc. 78 (23): 6208. doi:10.1021/ja01604a072.
- ↑ Krimm, H. (1957). Chem. Abstr. 256.
- ↑ Horner, L.; Jürgens, E. (1957). "Notiz Über Darstellung und Eigenschaften Einiger Isonitrone (Oxazirane)". Chemische Berichte 90 (10): 2184. doi:10.1002/cber.19570901010.
- ↑ 5.0 5.1 5.2 5.3 5.4 Davis, F. A.; Sheppard, A. C. (1989). "Applications of oxaziridines in organic synthesis". Tetrahedron 45 (18): 5703. doi:10.1016/s0040-4020(01)89102-x.
- ↑ Davis, F. A.; Stringer, O. D. (1982). "Chemistry of oxaziridines. 2. Improved synthesis of 2-sulfonyloxaziridines". The Journal of Organic Chemistry 47 (9): 1774. doi:10.1021/jo00348a039.
- ↑ Davis, F. A.; Chattopadhyay, S.; Towson, J. C.; Lal, S.; Reddy, T. (1988). "Chemistry of oxaziridines. 9. Synthesis of 2-sulfonyl- and 2-sulfamyloxaziridines using potassium peroxymonosulfate (oxone)". The Journal of Organic Chemistry 53 (9): 2087. doi:10.1021/jo00244a043.
- ↑ 8.0 8.1 8.2 8.3 Davis, F. A.; Chen, B. C. (1992). "Asymmetric hydroxylation of enolates with N-sulfonyloxaziridines". Chem. Rev. 92 (5): 919. doi:10.1021/cr00013a008.
- ↑ Davis, F. A.; Jenkins, R. H.; Awad, S. B.; Stringer, O. D.; Watson, W. H.; Galloy, J. (1982). "Chemistry of oxaziridines. 3. Asymmetric oxidation of organosulfur compounds using chiral 2-sulfonyloxaziridines". Journal of the American Chemical Society 104 (20): 5412. doi:10.1021/ja00384a028.
- ↑ Davis, F. A.; Reddy, R. T.; McCauley, J. P.; Przeslawski, R. M.; Harakal, M. E.; Carroll, P. J. (1991). "Chemistry of oxaziridines. 15. Asymmetric oxidations using 3-substituted 1,2-benzisothiazole 1,1-dioxide oxides". The Journal of Organic Chemistry 56 (2): 809. doi:10.1021/jo00002a056.
- ↑ 11.0 11.1 Towson, J. C.; Weismiller, M. C.; Lal, S. G.; Sheppard, A. C.; Davis, F. A. (1990). Org. Synth. 69: 158.
- ↑ Davis, F. A.; Towson, J. C.; Weismiller, M. C.; Lal, S.; Carroll, P. J. (1988). "Chemistry of oxaziridines. 11. (Camphorylsulfonyl)oxaziridine: synthesis and properties". Journal of the American Chemical Society 110 (25): 8477. doi:10.1021/ja00233a025.
- ↑ Bach, R. D.; Coddens, B. A.; McDouall, J. J. W.; Schlegel, H. B.; Davis, F. A. (1990). "The mechanism of oxygen transfer from an oxaziridine to a sulfide and a sulfoxide: a theoretical study". The Journal of Organic Chemistry 55 (10): 3325. doi:10.1021/jo00297a062.
- ↑ 14.0 14.1 Davis, F. A.; Kumar, A.; Chen, B. C. (1991). "Chemistry of oxaziridines. 16. A short, highly enantioselective synthesis of the AB-ring segments of .gamma.-rhodomycionone and .alpha.-citromycinone using (+)-[(8,8-dimethoxycamphoryl)sulfonyl]oxaziridine". The Journal of Organic Chemistry 56 (3): 1143. doi:10.1021/jo00003a042.
- ↑ Davis, F. A.; Weismiller, M. C.; Lal, G. S.; Chen, B. C.; Przeslawski, R. M. (1989). "(Camphorylsulfonyl)imine dianion in the synthesis of new optically pure (camphorylsulfonyl)oxaziridine derivatives". Tetrahedron Letters 30 (13): 1613. doi:10.1016/s0040-4039(00)99534-0.
- ↑ Chen, B. C.; Weismiller, M. C.; Davis, F. A.; Boschelli, D.; Empfield, J. R.; Smith, A. B. (1991). "Enantioselective synthesis of (+)-kjellmanianone". Tetrahedron 47 (2): 173–82. doi:10.1016/S0040-4020(01)80914-5.
- ↑ 17.0 17.1 17.2 Petrov, V. A.; Resnati, G. (1996). "Polyfluorinated Oxaziridines: Synthesis and Reactivity". Chemical Reviews 96 (5): 1809. doi:10.1021/cr941146h.
- ↑ 18.0 18.1 Evans, D. A.; Morrissey, M. M.; Dorow, R. L. (1985). "Asymmetric oxygenation of chiral imide enolates. A general approach to the synthesis of enantiomerically pure .alpha.-hydroxy carboxylic acid synthons". journal of the American Chemical Society 107 (14): 4346. doi:10.1021/ja00300a054.
- ↑ Dounay, Amy B.; Forsyth, Craig J. (1999). "Abbreviated Synthesis of the C3−C14 (Substituted 1,7-Dioxaspiro[5.5]undec-3-ene) System of Okadaic Acid". Org. Lett. 1 (3): 451. doi:10.1021/ol9906615.
- ↑ Malgesini, Beatrice; Forte, Barbara; Borghi, Daniela; Quartieri, Francesca; Gennari, Cesare; Papeo, Gianluca (2009). "A Straightforward Total Synthesis of (−)-Chaetominine". Chem. Eur. J. 15 (32): 7922. doi:10.1002/chem.200900793.
- ↑ Bohé, Luis; Hanquet, Gilles; Lusinchi, Marie; Lusinchi, Xavier (1993). "The stereospecific synthesis of a new chiral oxaziridinium salt". Tetrahedron Letters 34 (45): 7271. doi:10.1016/S0040-4039(00)79306-3.
- ↑ Arnone, Alberto; Foletto, Stefania; Metrangolo, Pierangelo; Pregnolato, Massimo; Resnati, Giuseppe (1999). "Highly Enantiospecific Oxyfunctionalization of Nonactivated Hydrocarbon Sites by Perfluoro-cis-2-n-butyl-3-n-propyloxaziridine". Org. Lett. 1 (2): 281. doi:10.1021/ol990594e.
- ↑ Schmitz, E.; Ohme, R. (1964). "Isomere Oxime mit Dreiringstruktur". Chem. Ber. 97 (9): 2521. doi:10.1002/cber.19640970916.
- ↑ 24.0 24.1 24.2 Andreae, S.; Schmitz, E. (1991). "ChemInform Abstract: Electrophilic Aminations with Oxaziridines". ChemInform 22 (46): 327. doi:10.1002/chin.199146339.
- ↑ 25.0 25.1 25.2 Aubé, Jeffrey (1997). "Oxiziridine rearrangements in asymmetric synthesis". Chemical Society Reviews 26 (4): 269. doi:10.1039/CS9972600269.
- ↑ Lattes, Armand; Oliveros, Esther; Riviere, Monique; Belzeck, Czeslaw; Mostowicz, Danuta; Abramskj, Wojciech;Piccinni-Leopardi, Carla; Germain, Gabriel; Van Meerssche, Maurice (1982). "Photochemical and thermal rearrangement of oxaziridines. Experimental evidence in support of the stereoelectronic control theory". Journal of the American Chemical Society 104 (14): 3929. doi:10.1021/ja00378a024.
- ↑ "Yohimbe: MedlinePlus Supplements". nlm.nih.gov. November 19, 2010. Retrieved December 13, 2010.