Onsager reciprocal relations
| Thermodynamics | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
 | ||||||||||||||
|
Branches |
||||||||||||||
|
History / Culture
|
||||||||||||||














In thermodynamics, the Onsager reciprocal relations express the equality of certain ratios between flows and forces in thermodynamic systems out of equilibrium, but where a notion of local equilibrium exists.
"Reciprocal relations" occur between different pairs of forces and flows in a variety of physical systems. For example, consider fluid systems described in terms of temperature, matter density, and pressure. In this class of systems, it is known that temperature differences lead to heat flows from the warmer to the colder parts of the system; similarly, pressure differences will lead to matter flow from high-pressure to low-pressure regions. What is remarkable is the observation that, when both pressure and temperature vary, temperature differences at constant pressure can cause matter flow (as in convection) and pressure differences at constant temperature can cause heat flow. Perhaps surprisingly, the heat flow per unit of pressure difference and the density (matter) flow per unit of temperature difference are equal. This equality was shown to be necessary by Lars Onsager using statistical mechanics as a consequence of the time reversibility of microscopic dynamics (microscopic reversibility). The theory developed by Onsager is much more general than this example and capable of treating more than two thermodynamic forces at once, with the limitation that "the principle of dynamical reversibility does not apply when (external) magnetic fields or Coriolis forces are present", in which case "the reciprocal relations break down".[1]
Though the fluid system is perhaps described most intuitively, the high precision of electrical measurements makes experimental realisations of Onsager's reciprocity easier in systems involving electrical phenomena. In fact, Onsager's 1931 paper[1] refers to thermoelectricity and transport phenomena in electrolytes as well-known from the 19th century, including "quasi-thermodynamic" theories by Thomson and Helmholtz respectively. Onsager's reciprocity in the thermoelectric effect manifests itself in the equality of the Peltier (heat flow caused by a voltage difference) and Seebeck (electrical current caused by a temperature difference) coefficients of a thermoelectric material. Similarly, the so-called "direct piezoelectric" (electrical current produced by mechanical stress) and "reverse piezoelectric" (deformation produced by a voltage difference) coefficients are equal. For many kinetic systems, like the Boltzmann equation or chemical kinetics, the Onsager relations are closely connected to the principle of detailed balance[1] and follow from them in the linear approximation near equilibrium.
Experimental verifications of the Onsager reciprocal relations were collected and analyzed by D.G. Miller[2] for many classes of irreversible processes, namely for thermoelectricity, electrokinetics, transference in electrolytic solutions, diffusion, conduction of heat and electricity in anisotropic solids, thermomagnetism and galvanomagnetism. In this classical review, chemical reactions are considered as "cases with meager" and with inconclusive evidence. Further theoretical analysis and experiments support the reciprocal relations for chemical kinetics with transport.[3]
For his discovery of these reciprocal relations, Lars Onsager was awarded the 1968 Nobel Prize in Chemistry. The presentation speech referred to the three laws of thermodynamics and then added "It can be said that Onsager's reciprocal relations represent a further law making a thermodynamic study of irreversible processes possible."[4] Some authors have even described Onsager's relations as the "Fourth law of thermodynamics".[5]
Example: Fluid system
The fundamental equation
The basic thermodynamic potential is internal energy. In a simple fluid system, neglecting the effects of viscosity the fundamental thermodynamic equation is written:
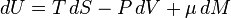
where U is the internal energy, T is temperature, S is entropy, P is the hydrostatic pressure,  is the chemical potential, and M mass. In terms of the internal energy density, u, entropy density s, and mass density
is the chemical potential, and M mass. In terms of the internal energy density, u, entropy density s, and mass density  , the fundamental equation is written:
, the fundamental equation is written:
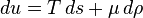
For non-fluid or more complex systems there will be a different collection of variables describing the work term, but the principle is the same. The above equation may be solved for the entropy density:
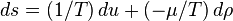
The above expression of the first law in terms of entropy change defines the entropic conjugate variables of  and , which are
and , which are 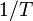 and
and 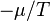 and are intensive quantities analogous to potential energies; their gradients of are called thermodynamic forces as they cause flows of the corresponding extensive variables as expressed in the following equations.
and are intensive quantities analogous to potential energies; their gradients of are called thermodynamic forces as they cause flows of the corresponding extensive variables as expressed in the following equations.
The continuity equations
The extensive quantities  and
and  are conserved and their flows satisfy continuity equations. The conservation of mass is written:
are conserved and their flows satisfy continuity equations. The conservation of mass is written:

and, assuming that fluid velocity makes a negligible contribution to the energy flow, the conservation of energy is simply the conservation of the internal energy:
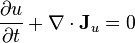
where 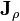 is the mass flux vector and
is the mass flux vector and 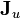 is the heat flux vector.
is the heat flux vector.
The entropy is not conserved and its continuity equation is written:
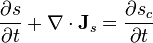
where 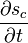 is the rate of increase in entropy density due to the irreversible processes of equilibration occurring in the fluid.
is the rate of increase in entropy density due to the irreversible processes of equilibration occurring in the fluid.
The phenomenological equations
In the absence of matter flows, Fourier's law is usually written:
-
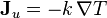 ;
;
where k is the thermal conductivity. However, this law is just a linear approximation, and holds only for the case where  , with the thermal conductivity possibly being a function of the thermodynamic state variables, but not their gradients or time rate of change. Assuming that this is the case, Fourier's law may just as well be written:
, with the thermal conductivity possibly being a function of the thermodynamic state variables, but not their gradients or time rate of change. Assuming that this is the case, Fourier's law may just as well be written:
-
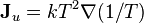 ;
;
In the absence of heat flows, Fick's law of diffusion is usually written:
-
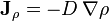 ,
,
where D is the coefficient of diffusion. Since this is also a linear approximation and since the chemical potential is monotonically increasing with density at a fixed temperature, Fick's law may just as well be written:
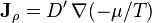
where, again, D' is a function of thermodynamic state parameters, but not their gradients or time rate of change. For the general case in which there both mass and energy fluxes, the phenomenological equations may be written as:
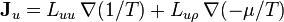
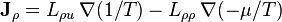
or, more concisely,

where the entropic "thermodynamic forces" conjugate to the "displacements" and are  and
and  and
and 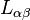 is the Onsager matrix of phenomenological coefficients.
is the Onsager matrix of phenomenological coefficients.
The rate of entropy production
From the fundamental equation, it follows that:

and

Using the continuity equations, the rate of entropy production may now be written:

and, incorporating the phenomenological equations:
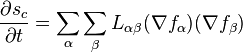
It can be seen that, since the entropy production must be greater than zero, the Onsager matrix of phenomenological coefficients is a positive semi-definite matrix.
The Onsager reciprocal relations
Onsager's contribution was to demonstrate that not only is positive semi-definite, it is also, except in certain special cases, symmetric. In other words, the cross-coefficients 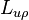 and
and 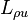 are equal. The fact that they are at least proportional follows from simple dimensional analysis (i.e., both coefficients are measured in the same units of temperature times mass density).
are equal. The fact that they are at least proportional follows from simple dimensional analysis (i.e., both coefficients are measured in the same units of temperature times mass density).
The rate of entropy production for the above simple example uses only two entropic forces, and a 2x2 Onsager phenomenological matrix. The expression for the linear approximation to the fluxes and the rate of entropy production can very often be expressed in an analogous way for many more general and complicated systems.
Abstract formulation
Let 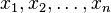 denote fluctuations from equilibrium values in several thermodynamic quantities, and let
denote fluctuations from equilibrium values in several thermodynamic quantities, and let 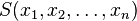 be the entropy. Then, Boltzmann's entropy formula gives for the probability distribution function
be the entropy. Then, Boltzmann's entropy formula gives for the probability distribution function 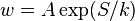 , A=const, since the probability of a given set of fluctuations
, A=const, since the probability of a given set of fluctuations 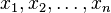 is proportional to the number of microstates with that fluctuation. Assuming the fluctuations are small, the probability distribution function can be expressed through the second differential of the entropy[6]
is proportional to the number of microstates with that fluctuation. Assuming the fluctuations are small, the probability distribution function can be expressed through the second differential of the entropy[6]
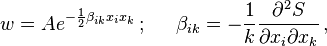
where we are using Einstein summation convention and  is a positive definite symmetric matrix.
is a positive definite symmetric matrix.
Using the quasi-stationary equilibrium approximation, that is, Assuming that the system is only slightly non-equilibrium, we have[6] 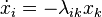
Suppose we define thermodynamic conjugate quantities as 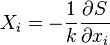 , which can also be expressed as linear functions (for small fluctuations):
, which can also be expressed as linear functions (for small fluctuations): 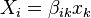
Thus, we can write  where
where 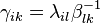 are called kinetic coefficients
are called kinetic coefficients
The principle of symmetry of kinetic coefficients or the Onsager's principle states that  is a symmetric matrix, that is
is a symmetric matrix, that is  [6]
[6]
Proof
Define mean values  and
and  of fluctuating quantities
of fluctuating quantities  and
and  respectively such that they take given values
respectively such that they take given values  at
at 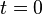 Note that
Note that 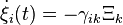
Symmetry of fluctuations under time reversal implies that 
or, with , we have 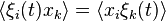
Differentiating with respect to 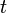 and substituting, we get
and substituting, we get 
Putting in the above equation, 
It can be easily shown from the definition that 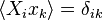 , and hence, we have the required result.
, and hence, we have the required result.
See also
References
- ↑ 1.0 1.1 1.2 L. Onsager, Reciprocal Relations in Irreversible Processes. I., Phys. Rev. 37, 405 - 426 (1931)
- ↑ D.G. Miller, Thermodynamics of irreversible processes. The experimental verification of the Onsager reciprocal relations, Chem. Rev. 60 (1960), 15-37.
- ↑ G.S. Yablonsky, A.N. Gorban, D. Constales, V.V. Galvita and G.B. Marin, Reciprocal relations between kinetic curves, EPL, 93 (2011) 20004.
- ↑ The Nobel Prize in Chemistry 1968. Presentation Speech.
- ↑ For example Richard P. Wendt, Journal of Chemical Education v.51, p.646 (1974) "Sîmplified Transport Theory for Electrolyte Solutions"
- ↑ 6.0 6.1 6.2 Landau, L. D.; Lifshitz, E.M. (1975). Statistical Physics, Part 1. Oxford, UK: Butterworth-Heinemann. ISBN 978-81-8147-790-3.