Idiopathic pulmonary fibrosis
Idiopathic pulmonary fibrosis (IPF) is a chronic and ultimately fatal disease characterized by a progressive decline in lung function.[2][3] The term pulmonary fibrosis means scarring of lung tissue and is the cause of worsening dyspnea (shortness of breath). Fibrosis is usually associated with a poor prognosis.[2][3][4] The term 'idiopathic' is used because the cause of pulmonary fibrosis is still unknown.[2]
IPF usually occurs in adult individuals of between 50 and 70 years of age, particularly those with a history of cigarette smoking, and affects more men than women.[2][5]
IPF belongs to a large group of more than 200 lung diseases known as interstitial lung diseases (ILD), characterized by the involvement of lung interstitium.[3] The interstitium, the tissue between the air sacs in the lung, is the primary site of injury in ILDs. However, these disorders frequently affect not only the interstitium, but also the airspaces, peripheral airways, and vessels.[3] Lung tissue from people with IPF shows a characteristic histopathologic pattern known as usual interstitial pneumonia (UIP). UIP is therefore the pathologic counterpart of IPF.[2][5]
In 2011, new guidelines for the diagnosis and management of IPF were published.[2] The diagnosis of IPF requires exclusion of other known causes of ILD and the presence of a typical radiological pattern identified through high resolution computed tomography (HRCT). In the right clinical setting, it is possible to make the diagnosis of IPF by HRCT alone, obviating the need for surgical lung biopsy.[2][3]
Classification
IPF belongs to as interstitial lung diseases which are characterized by the involvement of the lung interstitium,[3] the tissue between the air sacs of the lung. IPF is one specific presentation of idiopathic interstitial pneumonia (IIP), which is in turn a type of ILD, also known as diffuse parenchymal lung disease (DPLD).
The most important distinction among the IIPs is that between IPF and the other IIPs.[3] IIPs consist of disorders of known, as well as disorders of unknown causes. Other forms of IIPs include non-specific interstitial pneumonia (NSIP), desquamative interstitial pneumonia (DIP) and acute interstitial pneumonia (AIP).[3]
| Idiopathic pulmonary fibrosis | |
|---|---|
| Classification and external resources | |
| ICD-10 | J84.1 |
| ICD-9 | 516.3 |
| OMIM | 178500 |
| DiseasesDB | 4815 |
| MedlinePlus | 000069 |
| eMedicine | radio/873 |
| MeSH | D011658 |
Examples of ILD of known cause include hypersensitivity pneumonitis, pulmonary Langerhan’s cell histiocytosis, asbestosis and collagen vascular disease. However, these disorders frequently affect not only the interstitium, but also the airspaces, peripheral airways, and blood vessels.[3]
A new classification of ILDs, with a new allocation of the diseases, will be published soon.
Epidemiology
Although rare, IPF is the most common form of IIP.[3] It is estimated that in the USA, IPF affects between 132,000–500,000 people.[5] The prevalence of IPF has been estimated between 14.0 and 42.7 per 100,000 persons based on a USA analysis of healthcare claims data, with variation depending on the case definitions used in this analyses.[5][6] IPF is more common in men than in women and is usually diagnosed in people over 50 years of age.[2]
The incidence of IPF, estimated at approximately 50,000 new cases diagnosed annually,[6] is difficult to determine as uniform diagnostic criteria have not been applied consistently.[2] In the 27 European Union countries, a range of sources estimate an incidence of 4.6–7.4 people per 100,000 of the population,[7][8] suggesting that approximately 30,000–35,000 new patients will be diagnosed with IPF each year.[5][9]
Etiology and pathobiology
Despite extensive investigation, the cause of IPF remains unknown.[2] The fibrosis in IPF has been linked to cigarette smoking, environmental factors (e.g. occupational exposure to gases, smoke, chemicals or dusts), other medical conditions including gastroesophageal reflux disease (GERD), or to genetic predisposition (familial IPF). However, none of these is present in all people with IPF and therefore do not provide a completely satisfactory explanation for the disease.[2][10]
IPF is believed to be the result of an aberrant wound healing process including/involvingabnormal and excessive deposition of collagen(fibrosis) in the pulmonary interstitium with minimal associated inflammation.[11]
It is hypothesized that the initial or repetitive injury in IPF occurs to the lung cells, called alveolar epithelial cells (AECs, pneumocytes), which line the majority of the alveolar surface.[12] When type I AECs are damaged or lost, it is thought that type II AECs undergo proliferation to cover the exposed basement membranes. In normal repair, the hyperplastic type II AECs die and the remaining cells spread and undergo a differentiation process to become type I AECs. Under pathologic conditions and in the presence of transforming growth factor beta (TGFβ), fibroblasts accumulate in these areas of damage and differentiate into myofibroblasts that secrete collagen and other proteins.[12] In the past, It was thought that inflammation was the first event in initiating lung tissue scarring. According to the most recent findings, however, the development of fibroblastic foci precedes the accumulation of inflammatory cells and the consequent deposition of collagen.[13]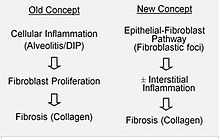
Familial IPF accounts for less than 5% of the total of patients with IPF and is clinically and histologically indistinguishable from sporadic IPF.[2] Genetic associations include mutations in pulmonary surfactant proteins A1, A2, C (SFTPA1, SFTPA2B) and mucin (MUC5B).[15] Mutations in human telomerase genes are also associated with familial pulmonary fibrosis and in some patients with sporadic IPF (TERT, TERC).[15]
Diagnosis
An earlier diagnosis of IPF is a prerequisite for earlier treatment and, potentially, improvement of the long-term clinical outcome of this progressive and ultimately fatal disease.[2] If IPF is suspected, diagnosis can be challenging but a multidisciplinary approach involving a pulmonologist, radiologist and pathologist expert in interstitial lung disease has been shown to improve the accuracy of IPF diagnosis.[2][16][17]
A Multidisciplinary Consensus Statement on the Idiopathic Interstitial Pneumonias published by the American Thoracic Society (ATS) and the European Respiratory Society (ERS) in 2000 proposed specific major and minor criteria for establishing the diagnosis of IPF.[3] However, in 2011, new simplified and updated criteria for the diagnosis and management of IPF were published by the ATS, ERS, together with the Japanese Respiratory Society (JRS) and Latin American Thoracic Association (ALAT).[2] Currently, a diagnosis of IPF requires:
- Exclusion of known causes of ILD, e.g., domestic and occupational environmental exposures, connective tissue disorders, or drug exposure/toxicity
- The presence of a typical radiological UIP pattern on HRCT.
In the right clinical setting, it is possible to make the diagnosis of IPF by HRCT alone, obviating the need for surgical lung biopsy.[2][3]
Recognizing IPF in clinical practice can be challenging as symptoms often appear similar to those of more common diseases, such asthma, chronic obstructive pulmonary disease (COPD) and congestive heart failure. The key issue facing clinicians is whether the presenting history, symptoms (or signs), radiology, and pulmonary function testing are collectively in keeping with the diagnosis of IPF or whether the findings are due to another process. It has long been recognized that patients with ILD related to asbestos exposure, drugs (such as chemotherapeutic agents or nitrofurantoin), rheumatoid arthritis and scleroderma/systemic sclerosis may be difficult to distinguish from IPF. Other differential diagnostic considerations include interstitial lung disease related to mixed connective tissue disease, advanced sarcoidosis, chronic hypersensitivity pneumonitis, pulmonary Langerhan’s cell histiocytosis and radiation-induced lung injury.[2][3]
Clinical features

In many patients, symptoms are present for a considerable time before diagnosis.[4] The most common clinical features of IPF include:[2][3][18]
- Age over 50 years
- Dry, non-productive cough on exertion
- Progressing exertional dyspnea (difficulty breathing on exercise)
- Dry, inspiratory bibasilar “velcro-like” crackles on auscultation (a crackling sound in the lungs during inhalation similar to Velcro being torn apart slowly, heard with a stethoscope).[2][6][19]
- Clubbing of the digits, a disfigurement of the finger tips or toes (see image)
- Abnormal pulmonary function test results, with evidence of restriction and impaired gas exchange.
These features are due to chronic oxygen deficiency in blood and can occur in a wide variety of other pulmonary disorders and not specific for IPF. However, IPF should be considered in all patients with unexplained chronic exertional dyspnea who present with cough, inspiratory bibasilar crackles, or finger clubbing.[2]
Assessment of “velcro” crackles on lung auscultation is a practical way to improve the earlier diagnosis of IPF. Fine crackles are easily recognized by clinicians and are characteristic of IPF.[20]
 |
IPF Lung Sound
velcro crackles on auscultation in a patient with IPF
|
| Problems playing this file? See media help. | |
If bilateral fine crackles are present throughout the inspiratory time and are persisting after several deep breaths, and if remaining present on several occasions several weeks apart in a subject aged ≥60 years, this should raise the suspicion of IPF and lead to consideration of an HRCT scan of the chest which is more sensitive than a chest X-ray.[19] As crackles are not specific for IPF, they must prompt a thorough diagnostic process.[2]
Recognition of these symptoms requires appropriate further investigations for diagnosing IPF.
Radiology
Chest X-rays is useful in the follow up routine of IPF patients. Plain chest X-rays are unfortunately not diagnostic but may reveal decreased lung volumes, typically with prominent reticular interstitial markings near the lung bases.[3]
(1–2 mm). Typical HRCT of the chest of IPF demonstrates fibrotic changes in both lungs, with a predilection for the bases and the periphery. According to the joint ATS/ERS/JRS/ALAT 2011 guidelines, HRCT is an essential component of the diagnostic pathway in IPF which can identify UIP by the presence of:[2]
- Reticular opacities, often associated with traction bronchiectasis
- Honeycombing manifested as cluster cystic airspaces, typically of comparable diameters (3–10 mm) but occasionally large. Usually sub-pleural and characterized by well-defined walls and disposed in at least two lines. Generally one line of cysts is not sufficient to define honeycombing
- Ground-glass opacities are common but less extensive than the reticulation
- Distribution characteristically basal and peripheral though often patchy.

Histology
According to the updated 2011 guidelines, in the absence of a typical UIP pattern on HRCT, a surgical lung biopsy is required for confident diagnosis.[2]
Histologic specimens for the diagnosis of IPF must be taken at least in three different places and be large enough that the pathologist can comment on the underlying lung architecture. Small biopsies, such as those obtained via transbronchial lung biopsy (performed during bronchoscopy) are usually not sufficient for this purpose. Hence, larger biopsies obtained surgically via a thoracotomy or thoracoscopy are usually necessary.[3]
Lung tissue from people with IPF usually show a characteristic histopathologic UIP pattern and is therefore the pathologic counterpart of IPF.[2][5] Although a pathologic diagnosis of UIP often corresponds to a clinical diagnosis of IPF, a UIP histologic pattern can be seen in other diseases as well, and fibrosis of known origin (rheumatic diseases for example).[2][3] There are four key features of UIP including interstitial fibrosis in a ‘patchwork pattern’, interstitial scarring, honeycomb changes and fibroblast foci.
Fibroblastic foci are dense collections of myofibroblasts and scar tissue and, together with honeycombing, are the main pathological findings that allow a diagnosis of UIP.

Bronchoalveolar lavage
Bronchoalveolar lavage (BAL) is a well-tolerated diagnostic procedure in ILD.[21] BAL cytology analyses (differential cell counts) should be considered in the evaluation of patients with IPF at the discretion of the treating physician based on availability and experience at their institution. BAL may reveal alternative specific diagnoses: malignancy, infections, eosinophilic pneumonia, histiocytosis X, or alveolar proteinosis. In the evaluation of patients with suspected IPF, the most important application of BAL is in the exclusion of other diagnoses. Prominent lymphocytosis (>40%) should suggest the diagnosis of IPF.[2]
Pulmonary function tests
Spirometry classically reveals a reduction in the vital capacity (VC) with either a proportionate reduction in airflows, or increased airflows for the observed vital capacity. The latter finding reflects the increased lung stiffness (reduced lung compliance) associated with pulmonary fibrosis, which leads to increased lung elastic recoil.[22]
Measurement of static lung volumes using body plethysmography or other techniques typically reveals reduced lung volumes (restriction). This reflects the difficulty encountered in inflating the fibrotic lungs.
The diffusing capacity for carbon monoxide (DLCO) is invariably reduced in IPF and may be the only abnormality in mild or early disease. Its impairment underlies the propensity of patients with IPF to exhibit oxygen desaturation with exercise which can also be evaluated using the 6-minute walk test (6MWT).[2]
Terms such as ‘mild’, ‘moderate’, and ‘severe’ are sometimes used for staging disease and are commonly based on resting pulmonary function test measurements.[2] However, there is no clear consensus regarding the staging of IPF patients and what are the best criteria and values to use. Mild-to-moderate IPF has been characterized by the following functional criteria:[23][24]
- Forced Vital Capacity (FVC) of ≥50%
- DLCO of ≥35%
- 6MWT distance ≥150 meters.
Prognosis

The clinical course of IPF can be unpredictable.[2][25] IPF progression is associated with an estimated median survival time of 2 to 5 years following diagnosis.[2][3] The 5-year survival for IPF ranges between 20–40%,[24] a mortality rate higher than that of a number of malignancies, including colon cancer, multiple myeloma and bladder cancer.[1][25]
Recently a multidimensional index and staging system has been proposed to predict mortality in IPF.[26] The name of the index is GAP and is based on gender [G], age [A], and two lung physiology variables [P] (FVC and DLCO] that are commonly measured in clinical practice to predict mortality in IPF. The highest stage of GAP (stage III) has been found to be associated with a 39% risk of mortality at 1 year.[26]
Treatment
The goals of treatment in IPF are essentially to reduce the symptoms, stop disease progression, prevent acute exacerbations, and prolong survival. Preventive care (e.g. vaccinations) and symptom-based treatment should be started early in every patient.[27]
Pharmacologic interventions
- N-acetylcysteine and triple therapy
N-Acetylcysteine (NAC) is a precursor to glutathione, an antioxidant. It has been hypothesized that treatment with high doses of NAC may repair an oxidant–antioxidant imbalance that occurs in the lung tissue of patients with IPF. In the first clinical trial of 180 patients (IFIGENIA), NAC was shown to reduce the decline in VC and DLCO over 12 months of follow-up when used in combination with prednisone and azathioprine (triple therapy).[28]
More recently, a large randomized, controlled trial (PANTHER-IPF) was undertaken by the National Institutes of Health (NIH) in the USA to evaluate triple therapy and NAC monotherapy in IPF patients. This study found that the combination of prednisone, azathioprine, and NAC increased the risk of death and hospitalizations and the NIH announced in 2012 that the triple-therapy arm of the PANTHER-IPF study had been terminated early.[29] This study is also evaluating NAC alone and the results are expected in 2013 (see below).
Considering these limitations, neither triple therapy nor NAC monotherapy are currently recommended for the majority of patients with IPF in the most recent IPF guidelines.[2][30]
- Anti-fibrotic agents
A number of treatments have been investigated in the past for IPF, including interferon gamma-1β,[31] bosentan,[32] ambrisentan,[33] and anticoagulants,[34] but these are no longer considered effective treatment options. Many of these earlier studies were based on the hypothesis that IPF is an inflammatory disorder.
Pirfenidone, however, is a small molecule that combines both anti-inflammatory, anti-oxidant, and anti-fibrotic effects in experimental models of fibrosis.[35] Pirfenidone, marketed under the trade name Esbriet, is approved in Europe for the treatment of patients with mild-to-moderate IPF. It is also approved in Japan (trade name Pirespa), India and China.
Pirfenidone was approved in the European Union based on the results of three Phase III, randomized, double-blind, placebo-controlled studies, one conducted in Japan and the other two in Europe and the USA (CAPACITY trials).[23][36]
A Review on the Cochrane Library (the journal of the Cochrane Collaboration for evidence-based Medicine) based on four trials involving 1155 patients comparing pirfenidone with placebo, demonstrated a significantly reduced risk of disease progression in patients treated with pirfenidone by 30%.[37] FVC or VC was also significantly improved by pirfenidone, even if a mild slowing in FVC decline could be demonstrated only in one of the two CAPACITY trials.[12] On the basis of these mixed results, the US Food and Drug Administration (FDA) demanded a third Phase III clinical study, ASCEND (NCT01366209), currently underway in the USA.[38]
- Future therapeutic options
A number of targeted therapies are currently in early testing or are being considered for development. These molecules are directed against several growth factors and cytokines that are known to play a role in the proliferation, activation, differentiation or inappropriate survival of fibroblasts.
Other current treatments being evaluated in advanced clinical trials for IPF comprise:
- Nintedanib (BIBF 1120) is being studied in two Phase III trials scheduled to be completed in late 2013.[39][40]
- N-acetylcysteine monotherapy is being studied in the PANTHER-IPF trial[41]
- A Phase II study of STX-100 is ongoing.[42]
More information can be found at ClinicalTrials.gov, a registry and results database of publicly and privately supported clinical studies of human participants conducted around the world.
Non pharmacological interventions
- Lung transplantation
Lung transplantation may be suitable for those patients physically eligible to undergo a major transplant operation. In IPF patients, lung transplant has been shown to reduce the risk of death by 75% as compared with patients who remain on the waiting list.[43] Since the introduction of the lung allocation score (LAS), which prioritizes transplant candidates based on survival probability, IPF has become the most common indication for lung transplantation in the USA.[44]
Symptomatic patients with IPF younger than 65 years of age and with a body mass index (BMI) ≤26 kg/m2 should be referred for lung transplantation, but there are no clear data to guide the precise timing for LTx. Although controversial, the most recent data suggest that bilateral lung transplantation is superior to single lung transplantation in patients with IPF.[45] Five-year survival rates after lung transplantation in IPF are estimated at between 50 to 56%.[2][46][47]
- Long-term oxygen therapy (LTOT)
In the 2011 IPF guidelines, oxygen therapy, or supplementary oxygen for home use, became a strong recommendation for use in those patients with clinically significant resting hypoxemia. Although oxygen therapy has not been shown to improve survival in IPF, some data indicate an improvement in exercise capacity.[2][48]
- Pulmonary rehabilitation
Fatigue and loss of muscular mass are common and disabling problems for patients with IPF. Pulmonary rehabilitation may alleviate the overt symptoms of IPF and improve functional status by stabilizing and/or reversing the extrapulmonary features of the disease.[44][49] Typical programs of rehabilitation include exercise training, nutritional modulation, occupational therapy, education and psychosocial counseling. In late the phase of disease, IPF patients tend to discontinue physical activity due to increasing dyspnea. Whenever possible, this should be discouraged.
Palliative care
Palliative care focuses on reducing symptoms and improving the comfort of patients rather than treating the disease. This may include treatment of worsening symptoms with the use of chronic opioids for severe dyspnea and cough. Further, oxygen therapy may be useful for palliation of dyspnea in hypoxemic patients.
Palliative care also includes relief of physical and emotional suffering and psychosocial support for patients and caregivers.[2] With disease progression, patients may experience fear, anxiety and depression and psychological counseling should therefore be considered. In a recent study of outpatients with ILDs, including IPF, depression score, functional status (as assessed by walk test), as well as pulmonary function, all contributed to the severity of dyspnea.[50]
In selected cases of particularly severe dyspnea morphine could be considered. It can reduce dyspnea, anxiety and cough without significant decrease in oxygen saturation.[51]
Management and follow-up
IPF is often misdiagnosed, at least until physiological and/or imaging data suggest the presence of an ILD leading to delay in accessing appropriate care.[44] Considering that IPF is a disease with a median survival of three years after diagnosis, early referral to a center with specific expertise should therefore be considered for any patient with suspected or known ILD. On the basis of the complex differential diagnostic, multidisciplinary discussion between pulmonologists, radiologists, and pathologists experienced in the diagnosis of ILD is of the utmost importance to an accurate diagnosis.[2]
After diagnosis of IPF, and the appropriate treatment choice according to symptoms and stage of disease, a close follow-up should be applied. Due to the high variable course of disease, the higher incidence of complications such as lung cancer (up to 25% of patients has been reported in IPF) a routine evaluation every 3 to 6 months, including spirometry (body plethysmography), diffusion capacity testing, chest X-rays, 6MWT, assessment of dyspnea, quality of life, oxygen requirement is mandatory.
In addition, the increasing awareness of complications and common concomitant conditions frequently associated with IPF requires a routinely evaluation of comorbidities, most of them simply reflecting concurrent diseases of aging, and medications with their interaction and side effects.
Acute exacerbations
Acute exacerbations of IPF (AE-IPF) are defined as an unexplained worsening or development of dyspnea within 30 days with new radiological infiltrates at HRCT abnormality often superimposed on a background consistent with UIP pattern. The yearly incidence of AE-IPF is between 10 and 15% of all patients. The prognosis of AE-IPF is poor, with mortality ranging from 78% to 96%.[52] Other causes of AE-IPF such as pulmonary embolism, congestive heart failure, pneumothorax, or infection need to be excluded. Pulmonary infection have to be ruled out by endotracheal aspirate or BAL.
Many patients experiencing acute deterioration require intensive care treatment, particularly when respiratory failure is associated with hemodynamic instability, significant co-morbidities or severe hypoxemia.[53] However, mortality during hospitalization is high.[53] Mechanical ventilation should be introduced only after carefully weighing the patient’s long-term prognosis and, whenever possible, the patient’s wishes. However, current guidelines discourage the use of mechanical ventilation in patients with respiratory failure secondary to IPF.[2]
Some well-known cases of IPF
- Marlon Brando, screen and stage actor
- Robert Goulet, singer and actor
- Evel Knievel, legendary stuntman
- Steve Gerber, co-creator of the satiric comic book character Howard the Duck
- James Doohan, actor in Star Trek television and film series.
In other species
IPF has been recognized in several breeds of both dogs and cats,[54] and has been best characterized in West Highland White Terriers.[55] Veterinary patients with the condition share many of the same clinical signs as their human counterparts, including progressive exercise intolerance, increased respiratory rate, and eventual respiratory distress.[56] Prognosis is generally poor.
References
- ↑ 1.0 1.1 Bjoraker JA, Ryu JH, Edwin MK, Myers JL, Tazelaar HD, Schroeder DR, Offord KP (1998). "Prognostic significance of histopathologic subsets in idiopathic pulmonary fibrosis". Am J Respir Crit Care Med. 157 (1): 199–203. doi:10.1164/ajrccm.157.1.9704130. PMID 9445300.
- ↑ 2.0 2.1 2.2 2.3 2.4 2.5 2.6 2.7 2.8 2.9 2.10 2.11 2.12 2.13 2.14 2.15 2.16 2.17 2.18 2.19 2.20 2.21 2.22 2.23 2.24 2.25 2.26 2.27 2.28 2.29 2.30 2.31 2.32 2.33 2.34 2.35 Raghu G, Collard HR, Egan JJ, et al (2011). "An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management". Am. J Respir. Crit. Care Med. 183 (6): 788–824. doi:10.1164/rccm.2009-040GL. PMID 21471066.
- ↑ 3.0 3.1 3.2 3.3 3.4 3.5 3.6 3.7 3.8 3.9 3.10 3.11 3.12 3.13 3.14 3.15 3.16 3.17 American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. Am Respir Crit Care Med. 165 (2) 277–304. January 2002. PMID 11790668
- ↑ 4.0 4.1 Meltzer EB, Noble PW (2008). "Idiopathic pulmonary fibrosis". Orphanet J Rare Dis. 3 (1). doi:10.1186/1750-1172-3-8. PMC 2330030. PMID 18366757.
- ↑ 5.0 5.1 5.2 5.3 5.4 5.5 Pulmonary Fibrosis Foundation. "Prevalence and Incidence". Pulmonaryfibrosis.org. Retrieved 2013-04-11
- ↑ 6.0 6.1 6.2 Raghu G, Weycker D, Edesberg J, Bradford WZ, Oster G (2006). "Incidence and prevalence of idiopathic pulmonary fibrosis". Am. J Respir. Crit. Care Med 174 (7): 810–816.
- ↑ Gribbin J, Hubbard RB, Le Jeune I, Smith CJ, West J, Tata LJ (2006). "Incidence and mortality of idiopathic pulmonary fibrosis and sarcoidosis in the UK". Thorax 61 (11): 980–985. doi:10.1136/thx.2006.062836. PMC 2121155. PMID 16844727.
- ↑ Eurostat News Release. European demography. 110/2010. 27 July 2010
- ↑ García-Sancho C, Buendía-Roldán I, Fernández-Plata MR, Navarro C, Pérez-Padilla R, Vargas MH, Loyd JE, Selman M. Buendía-Roldán I, Fernández-Plata MR, et al. (2011). "Familial pulmonary fibrosis is the strongest risk factor for idiopathic pulmonary fibrosis". Respir Med. 105 (12): 1902–1990. doi:10.1016/j.rmed.2011.08.022. PMID 21917441.
- ↑ 11.0 11.1 Harari S, Caminati A (2010). "IPF: new insight on pathogenesis and treatment". Allergy 65 (5): 537–553. doi:10.1111/j.1398-9995.2009.02305. PMID 20121758.
- ↑ 12.0 12.1 12.2 Loomis-King H, Flaherty KR, Moore BB (April 2013). "Pathogenesis, current treatments and future directions for idiopathic pulmonary fibrosis". Current Opinion in Pharmacology 13 (3): 377–385. doi:10.1016/j.coph.2013.03.015.
- ↑ Pardo A, Selman M (2002). "Idiopathic pulmonary fibrosis: new insights in its pathogenesis". Int J Biochem Cell Biol. 34 (12): 1534–1538.
- ↑ Selman M, King TE, Pardo A (2001). "Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy". Annals of Internal Medicine 134 (2): 136–151. doi:10.7326/0003-4819-134-2-200101160-00015. PMID 11177318.
- ↑ 15.0 15.1 Online 'Mendelian Inheritance in Man' (OMIM) 178500
- ↑ Flaherty KR, King TE, Raghu G, Lynch JP, Colby TV, Travis WD, Gross BH, Kazerooni EA, et al. (2004). "Idiopathic interstitial pneumonia: what is the effect of a multidisciplinary approach to diagnosis?". Am J Respir Crit Care Med. 170 (8): 904–910. doi:10.1164/rccm.200402-147OC. PMID 15256390.
- ↑ Flaherty KR, Andrei AC, King TE Jr, Raghu G, Colby TV, Wells A, Bassily N, Brown K, et al. (2007). "Idiopathic interstitial pneumonia: do community and academic physicians agree on diagnosis?". Am J Respir Crit Care Med. 175 (10): 1054–1060. doi:10.1164/rccm.200606-833OC. PMC 1899268. PMID 17255566.
- ↑ Flaherty KR, Mumford JA, Murray S, Kazerooni EA, Gross BH, Colby TV, Travis WD, Flint A, et al. (2007). "Prognostic implications of physiologic and radiographic changes in idiopathic interstitial pneumonia". Am J Respir Crit Care Med. 168 (5): 543–548. doi:10.1164/rccm.200209-1112OC. PMID 12773329.
- ↑ 19.0 19.1 Cottin V, Cordier JF (2012). "Velcro crackles: the key for early diagnosis of idiopathic pulmonary fibrosis". Eur Respir J. 40 (3): 519–521. doi:10.1183/09031936.00001612. PMID 22941541.
- ↑ Baughman RP, Shipley RT, Loudon RG, Lower EE (1991). "Crackles in interstitial lung disease. Comparison of sarcoidosis and fibrosing alveolitis". Chest 100 (1): 96–101. PMID 2060395.
- ↑ Ohshimo S, Bonella F, Cui A, Beume M, Kohno N, Guzman J, Costabel U (2009). "Significance of bronchoalveolar lavage for the diagnosis of idiopathic pulmonary fibrosis". Am J Respir Crit Care Med. 179 (11): 1043–1047.
- ↑ Pellegrino R, Viegi G, Brusasco V, Crapo RO, Burgos F, Casaburi R, Coates A, van der Grinten CP, et al. (2005). "Interpretative strategies for lung function tests". Eur Respir J. 26 (5): 948–968. doi:10.1183/09031936.05.00035205. PMID 16264058.
- ↑ 23.0 23.1 Noble PW, Albera C, Bradford WZ, Costabel U, Glassberg MK, Kardatzke D, King TE, Lancaster L, et al. (2011). "Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials". Lancet 377 (9779): 1760–1769. doi:10.1016/S0140-6736(11)60405-4. PMID 21571362.
- ↑ Martinez FJ, Safrin S, Weycker D, Starko KM, Bradford WZ, King TE Jr, Flaherty KR, Schwartz DA, et al. IPF study group (2005). "The clinical course of patients with idiopathic pulmonary fibrosis". Annals of Internal Medicine 377 (9779): 1760–1769. doi:10.1016/S0140-6736(11)60405-4. PMID 21571362.
- ↑ 25.0 25.1 Kim DS, Collard HR, King TE (2006). "Classification and natural history of the idiopathic interstitial pneumonias". Proc Am Thorac Soc. 3 (4): 285–292. doi:10.1513/pats.200601-005TK. PMC 2658683. PMID 16738191.
- ↑ 26.0 26.1 Ley B, Ryerson CJ, Vittinghoff E, Ryu JH, Tomassetti S, Lee JS, Poletti V, Buccioli M, Elicker BM, Jones KD, King TE Jr, Collard HR. "A multidimensional index and staging system for idiopathic pulmonary fibrosis". Annals of Internal Medicine 156 (10): 684–691.
- ↑ Lee JS, McLaughlin S, Collard HR (2011). "Comprehensive care of the patient with idiopathic pulmonary fibrosis". Current Opinion in Pulmonary Medicine 17 (5): 348–354.
- ↑ Demedts M, Behr J, Buhl R, Costabel U, Dekhuijzen R, Jansen HM, MacNee W, Thomeer M, et al. (2005). "High-dose acetylcysteine in idiopathic pulmonary fibrosis". N Engl J Med. 353 (21): 2229–2242. doi:10.1056/NEJMoa042976. PMID 16306520.
- ↑ Commonly used three-drug regimen for idiopathic pulmonary fibrosis found harmful , Nih.gov., Retrieved 2013-04-11
- ↑ Behr J. (2012). "Prednisone, azathioprine an N-acetylcysteine for pulmonary fibrosis". N Engl J Med. 367 (9): 869–871. doi:10.1056/NEJMc1207471. PMID 22931324.
- ↑ King TE Jr, Albera C, Bradford WZ, Costabel U, Hormel P, Lancaster L, Noble PW, Sahn SA, et al. INSPIRE Study Group (2009). "Effect of interferon gamma-1b on survival in patients with idiopathic pulmonary fibrosis". Lancet 374 (9685): 222–228. doi:10.1016/S0140-6736(09)60551-1. PMID 19570573.
- ↑ King TE Jr, Brown KK, Raghu G, du Bois RM, Lynch DA, Martinez F, Valeyre D, Leconte I, et al. (2011). "BUILD-3: a randomized, controlled trial of bosentan in idiopathic pulmonary fibrosis". Am J Respir Crit Care Med. 184 (1): 92–99. doi:10.1164/rccm.201011-1874OC. PMID 21474646.
- ↑ Raghu G, Behr J, Brown KK, Egan JJ, Kawut SM, Flaherty KR, Martinez FJ, Nathan SD, et al. (2013). "Treatment of idiopathic pulmonary fibrosis with ambrisentan: a parallel, randomized trial". Annals of Internal Medicine 158 (9): 641–649. doi:10.7326/0003-4819-158-9-201305070-00003. PMID 23648946.
- ↑ Noth I, Anstrom KJ, Calvert SB, de Andrade J, Flaherty KR, Glazer C, Kaner RJ, Olman MA (2012). "Idiopathic Pulmonary Fibrosis Clinical Research Network (IPFnet) A placebo-controlled randomized trial of warfarin in idiopathic pulmonary fibrosis". Am J Respir Crit Care Med. 186 (1): 88–95. doi:10.1164/rccm.201202-0314OC. PMID 22561965.
- ↑ Schaefer CJ, Ruhrmund DW, Pan L, Seiwert SD, Kossen K (2011). "Antifibrotic activities of pirfenidone in animal models". Eur Respir Rev. 20 (120): 85–97.
- ↑ Taniguchi H, Ebina M, Kondoh Y, Ogura T, Azuma A, Suga M, Taguchi Y, Takahashi H, et al. (2010). "Pirfenidone in idiopathic pulmonary fibrosis". Eur Respir J. 35 (4): 821–829. doi:10.1183/09031936.00005209. PMID 19996196.
- ↑ Spagnolo P, Del Giovane C, Luppi F, Cerri S, Balduzzi S, Walters EH, D'Amico R, Richeldi L (2010). "Non-steroid agents for idiopathic pulmonary fibrosis". Cochrane Database Syst Rev. (9) CD003134. doi:10.1002/14651858.CD003134.pub2
- ↑ Efficacy and safety of pirfenidone in patients with idiopathic pulmonary fibrosis (IPF)(ASCEND). ClinicalTrials.gov. Retrieved 2013-04-11.
- ↑ Safety and Efficacy of BIBF 1120 at High Dose in Idiopathic Pulmonary Fibrosis Patients. ClinicalTrials.gov. Retrieved 2013-04-11.
- ↑ Safety and Efficacy of BIBF 1120 at High Dose in Idiopathic Pulmonary Fibrosis Patients II. ClinicalTrials.gov. Retrieved 2013-04-11.
- ↑ Evaluating the Effectiveness of Prednisone, Azathioprine, and N-acetylcysteine in Patients With IPF (PANTHER-IPF). ClinicalTrials.gov. Retrieved 2013-04-11.
- ↑ STX-100 in Patients With Idiopathic Pulmonary Fibrosis (IPF). ClinicalTrials.gov. Retrieved 2013-04-11.
- ↑ Russo MJ, Iribarne A, Hong KN, Davies RR, Xydas S, Takayama H, Ibrahimiye A, Gelijns AC, Bacchetta MD, D'Ovidio F, Arcasoy S, Sonett JR (2010). "High lung allocation score is associated with increased morbidity and mortality following transplantation". Chest 137 (3): 651–657.
- ↑ 44.0 44.1 44.2 Spagnolo P, Tonelli R, Cocconcelli E, Stefani A, Richeldi L (2012). "Idiopathic pulmonary fibrosis: diagnostic pitfalls and therapeutic challenges". Multidiscip Respir Med. 7 (1): 42.
- ↑ George TJ, Arnaoutakis GJ, Shah AS (2007). "Lung transplantation for idiopathic pulmonary fibrosis". Ann Thorac Surg. 84 (4): 1121–1128. doi:10.1016/j.athoracsur.2007.04.096. PMID 17888957.
- ↑ Mason DP, Brizzio ME, Alster JM, McNeill AM, Murthy SC, Budev MM, Mehta AC, Minai OA, et al. (2011). "Lung transplant in idiopathic pulmonary fibrosis". Arch Surg. 146 (10): 1204–1209.
- ↑ Keating D, Levvey B, Kotsimbos T, Whitford H, Westall G, Williams T, Snell G (2009). "Lung transplantation in pulmonary fibrosis challenging early outcomes counter balanced by surprisingly good outcomes beyond 15 years". Transplant Proc. 41 (1): 289–291. doi:10.1016/j.transproceed.2008.10.042. PMID 19249537.
- ↑ Morrison DA, Stovall JR. (1992). "Increased exercise capacity in hypoxemic patients after long-term oxygen therapy". Chest 102 (2): 542–550. PMID 1643945.
- ↑ Lee JS, McLaughlin S, Collard HR (2011). "Comprehensive care of the patient with idiopathic pulmonary fibrosis". Current Opinion in Pulmonary Medicine 17 (5): 348–354.
- ↑ Ryerson CJ, Berkeley J, Carrieri-Kohlman VL, Pantilat SZ, Landefeld CS, Collard HR (2011). "Depression and functional status are strongly associated with dyspnea in interstitial lung disease". Chest 139 (3): 609–616.
- ↑ Allen S, Raut S, Woollard J, Vassallo M (2005). "Low dose diamorphine reduces breathlessness without causing a fall in oxygen saturation in elderly patients with end-stage idiopathic pulmonary fibrosis". Palliat Med. 19 (2): 128–130.
- ↑ Agarwal R, Jindal SK (2008). "Acute exacerbation of idiopathic pulmonary fibrosis: a systematic review". Eur J Intern Med. 19 (4): 227–235.
- ↑ 53.0 53.1 Stern JB, Mal H, Groussard O, Brugière O, Marceau A, Jebrak G, Fournier M (2001). "Prognosis of patients with advanced idiopathic pulmonary fibrosis requiring mechanical ventilation for acute respiratory failure". Chest 120 (1): 213–219.
- ↑ Williams K, Malarkey D, Cohn L, Patrick D, Dye J, Toews G (2004). "Identification of spontaneous feline idiopathic pulmonary fibrosis: morphology and ultrastructural evidence for a type II pneumocyte defect". Chest 125 (6): 2278–2288. PMID 15189952.
- ↑ Webb JA, Armstrong J (2002). "Chronic idiopathic pulmonary fibrosis in a West Highland white terrier". Can Vet J. 43 (9): 703–705. PMC 339552. PMID 12240528.
- ↑ Canine Pulmonary Fibrosis. Akcchf.org. Retrieved 2013-04-11.
External links
| Wikimedia Commons has media related to Idiopathic pulmonary fibrosis. |
- Idiopathic pulmonary fibrosis on the Open Directory Project
- Idiopathic Pulmonary Fibrosis (IPF) Community
- Pulmonary fibrosis foundation
- AIR Community
- AIMIP Associazione Italiana Malattie Interstiziali o Rare del Polmone
- The European IPF Registry (eurIPFreg) has become Europe's leading database of longitudinal data from IPF patients, including control groups of patients with other lung diseases
- ILD CARE FOUNDATION´s activity is focused to increase knowledge, support research, contribute to prevention and provide counselling for interstitial lung diseases
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||