High-valent iron

High-valent iron commonly denotes compounds and intermediates in which iron is found in a formal oxidation state > 3 that show a number of bonds > 6 with a coordination number ≤ 6. The term is rather uncommon for hepta-coordinate compounds of iron.[1] It has to be distinguished from the terms hypervalent and hypercoordinate, as high-valent iron compounds neither necessarily violate the 18-electron rule nor necessarily show coordination numers > 6. The ferrate ion [FeO4]2- was the first structure in this class synthesized. The synthetic compounds discussed below contain highly oxidized iron in general, as the concepts are closely related.
Oxoiron compounds[2]
Oxoferryl species are commonly proposed as intermediates in catalytic cycles, especially biological systems in which O2 activation is required. Diatomic oxygen has a high reduction potential (E0 = 1.23 V), but the first step required to harness this potential is a thermodynamically unfavorable one electron reduction E0 = -0.16 V. This reduction occurs in nature by the formation of a superoxide complex in which a reduced metal is oxidized by O2. The product of this reaction is a peroxide radical that is more readily reactive. The abundance of these species in nature and the chemistry that is available to them are the reasons why the study of these compounds is important.[citation needed] A widely applicable method for the generation of high-valent oxoferryl species is the oxidation with iodosobenzene.
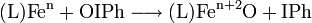
- symbolic oxidation of an iron compound using iodosobenzene, L denotes the supporting ligand
Fe(IV)O
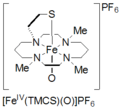
Several syntheses of oxoiron(IV) species have been reported. These compounds model biological complexes such as cytochrome P450, NO synthase, and isopenicillin N synthase. Two such reported compounds are thiolate-ligated oxoiron(IV) and cyclam-acetate oxoiron(IV). Thiolate-ligated oxoiron(IV) is formed by the oxidation of a precursor, [FeII(TMCS)](PF6) (TMCS = 1-mercaptoethyl-4,8,11-trimethyl-1,4,8,11-tetraza cyclotetradecane), and 3-5 equivalents of H2O2 at -60 ˚C in methanol. The iron(IV) compound is deep blue in color and shows intense absorption features at 460 nm, 570 nm, 850 nm, and 1050 nm. This species FeIV(=O)(TMCS)+ is stable at -60 ˚C, but decomposition is reported as temperature increases. Compound 2 was identified by Mössbauer spectroscopy, high resolution electrospray ionization mass spectrometry (ESI-MS), x-ray absorption spectroscopy, x-ray absorption fine structure (EXAFS), UV-visible spectroscopy (UV-vis), Fourier transform infrared spectroscopy (FT-IR), and results were compared to DFT-calculations.[3]
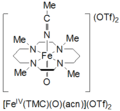
Tetramethylcyclam oxoiron(IV) is formed by the reaction of FeII(TMC)(OTf)2, TMC = 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane; OTf = CF3SO3, with iodosylbenzene (PhIO) in CH3CN at -40 ˚C. A second method for formation of cyclam oxoiron(IV) is reported as the reaction of FeII(TMC)(OTf)2 with 3 equivalents of H2O2 for 3 hours. This species is pale green in color and has an absorption maximum at 820 nm. It is reported to be stable for at least 1 month at -40 ˚C. It has been characterized by Mössbauer spectroscopy, ESI-MS, EXAFS, UV-vis, Raman Spectroscopy, and FT-IR.[4]
High-valent iron bispidine complexes are able to oxidize cyclohexane to cyclohexanol and cyclohexanone in 35% yield with an alcohol to ketone ratio up to 4.[5]
Fe(V)O
FeVTAML(=O), TAML = tetraamido macrocyclic ligand, is formed by the reaction of [FeIII(TAML)(H2O)](PPh4) with 2-5 equivalents of meta-chloroperbenzoic acid at -60 ˚C in n-butyronitrile. This compound has a deep green color with absorption maxima at 445 and 630 nm. It is reported to be stable for at least 1 month at 77 K. FeVTAML(=O) was characterized with electron paramagnetic resonance (EPR), Mössbauer, EXAFS, ESI-MS, reactivity studies, and DFT calculations. This compound is capable of 1 electron oxidation at the metal as opposed to at the ligand. This is due to the stronger –donor capacity of deprotonated amide nitrogens on TAML.[6]
Electronic structure
The electronic structure of porphyrin oxoiron compounds has been reviewed.[7]
Nitridoiron[8] & imidoiron[9] compounds

Nitrido- and imidoiron-compounds are closely related to iron-dinitrogen chemistry.[10] The biological significance of nitridoiron(V) porphyrins has been reviewed[11],.[12] A widely applicable method to generate high-valent nitridoiron species is the thermal or photochemical oxidative elimination of molecular nitrogen from an azide complex.
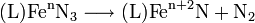
- symbolic oxidative elimination of nitrogen yields a nitridoiron complex, L denotes the supporting ligand
Fe(IV)N
Several structurally characterized nitridoiron(IV)-compounds exist [13],[14],.[15]
Fe(V)N
The first nitridoiron(V)-compound was generated and characterized in 1988/1989 by Wagner and Nakamoto using photolysis and Raman spectroscopy at low temperatures [16],.[17]
Fe(VI)N
A second FeVI species apart from the ferrate ion, [(Me3cy-ac)FeN](PF6)2, has been reported. This species, is formed by oxidation followed by photolysis to yield the Fe(VI) species. Characterization of the Fe(VI) complex was done by Mossbauer, EXAFS, IR, and DFT calculations. Unlike the ferrate ion compound 5 is diamagnetic.[18]
µ-nitrido compounds & oxidation catalysis[19]
Bridged µ-nitrido di-iron phthalocyanine compounds catalyze the oxidation of methane to methanol, formaldehyde and formic acid using hydrogen peroxide as sacrificial oxidant.[20]
Electronic structure
Nitridoiron(IV/V)-species were first explored theoretically in 2002.[21]
References
- ↑ Craig et al. Dalton Trans., 2010, 39, 4874-4881 doi:10.1039/B927032A
- ↑ Review: Que et al.; Journal of Inorganic Biochemistry Volume 100, Issue 4, April 2006, Pages 421-433;doi:10.1016/j.jinorgbio.2006.01.014
- ↑ Bukowski, M. R., Koehntop, K. D., Stubna, A., Bominaar E. L., Halfen, J. A., Munck, E., Nam, W., Que, L., Science, 310, 1000-1002, 2005; doi:10.1126/science.111909
- ↑ Rohde, J.-U., In, J.-H., Lim, M. H., Brennessel, W. W., Bikowski, M. R., Stubna, A., Munck, E., Name, W., Que, L., Science, 299, 1037-1039, 2003; doi:10.1126/science.299.5609.1037
- ↑ Comba, P. et al.; Inorg. Chem., 2009, 48 (21), pp 10389–10396; doi:10.1021/ic901702s
- ↑ Oliveira, F. T., Chanda, A., Banerjee, D., Shan, X., Mondal, S., Que, L., Bominaar, E. L., Munck, E., Collins, T. J., Science, 315, 835-838, 2007; doi: 10.1126/science.1133417
- ↑ Review: Fujii, H.; Coordination Chemistry Reviews Volume 226, Issues 1-2, March 2002, Pages 51-60; doi:10.1016/S0010-8545(01)00441-6
- ↑ Review: Berry, J.F.; Comments on Inorganic Chemistry, 30: 28–66, 2009; doi:10.1080/02603590902768875
- ↑ Review: Peters, J.C., Mehn, M.P.; Journal of Inorganic Biochemistry Volume 100, Issue 4, April 2006, Pages 634-643; doi:10.1016/j.jinorgbio.2006.01.023
- ↑ Review: Tyler, D. R., Crossland, J. E.; Coordination Chemistry Reviews 254 (2010) 1883–1894; doi:10.1016/j.ccr.2010.01.005
- ↑ Review: Nakamoto, K.; Coordination Chemistry Reviews Volume 226, Issues 1-2, March 2002, Pages 153-165; doi:10.1016/S0010-8545(01)00425-8
- ↑ Review: Nakamoto, K.; Journal of Molecular Structure Volumes 408-409, 1 June 1997, Pages 11-16; doi:10.1016/S0022-2860(96)09670-6
- ↑ Peters, Jonas C.; Que, Lawrence, Jr. et al.; Inorg. Chem., 2007, 46 (14), pp 5720–5726; doi:10.1021/ic700818q
- ↑ Smith et al.; Angewandte Chemie International Edition Volume 48, Issue 17, pages 3158–3160, April 14, 2009; doi:10.1002/anie.200900381
- ↑ Meyer et al.; Angewandte Chemie International Edition Volume 47, Issue 14, pages 2681–2684, March 25, 2008, April 14, 2009; doi:10.1002/anie.200800600
- ↑ Wagner, W.D.; Nakamoto, K.; J. Am. Chem. Soc., 1989, 111 (5), pp 1590–1598; doi:10.1021/ja00187a010
- ↑ Wagner, W.D.; Nakamoto, K.; J. Am. Chem. Soc., 1988, 110 (12), pp 4044–4045; doi:10.1021/ja00220a057
- ↑ Berry, J. F., Bill, E., Bothe, E., George, S. D., Miener, B., Neese, F., Wieghardt, K., Science, 312, 1937-1941, 2006; doi:10.1126/science.1128506
- ↑ Review: Que, L., Tolman, W.B.; Nature 455, 333-340 (18 September 2008); doi:10.1038/nature07371
- ↑ Sorokin, A.B.; Kudrik, E.V.; Bouchu, D.; Chem. Commun., 2008, 2562-2564; doi:10.1039/B804405H
- ↑ Dey, A.; Ghosh, A.; J. Am. Chem. Soc., 2002, 124 (13), pp 3206–3207; doi:10.1021/ja012402s
Further reading
- high-valent manganese: Jacobsen's catalyst
See also: Solomon et al.; Angewandte Chemie International Edition Volume 47, Issue 47, pages 9071–9074, November 10, 2008; doi:10.1002/anie.200803740