Exome sequencing

Exome sequencing (also known as targeted exome capture) is an efficient strategy to selectively sequence the coding regions of the genome as a cheaper but still effective alternative to whole genome sequencing. Exons are short, functionally important sequences of DNA which represent the regions in genes that are translated into protein. Exons are flanked by untranslated regions (UTR) that are usually not included in exome studies. In the human genome there are about 180,000 exons. These constitute about 1% of the human genome or about 30 megabases.[1]

The robust approach to sequencing the complete coding region (exome) has the potential to be clinically relevant in genetic diagnosis due to current understanding of functional consequences in sequence variation.[2] The goal of this approach is to identify the functional variation that is responsible for both mendelian and common diseases such as Miller syndrome and Alzheimer's disease without the high costs associated with whole-genome sequencing while maintaining high coverage in sequence depth.[2]
As an efficient strategy
There are many benefits to exome sequencing in the detection of rare causal variants of Mendelian disorders as opposed to whole genome sequencing or traditional linkage studies:
- Traditional genetic linkage studies which are followed by positional cloning require a large number of affected individuals. This approach is not suitable for rare Mendelian diseases because of the little amount of available cases. Linkage is furthermore not applicable to study affected individuals from different families or the cases caused by a new mutation not present in the parents.[3]
- Linkage studies are not robust enough to detect disorders with causal variants in different genes (genetic heterogeneity) or diseases that have diverse clinical features (phenotypic heterogeneity).[3]
- Because the human exome consists of 1% of the entire genome, it is possible to get deep coverage with relatively few reads.[3] Deep coverage is necessary for the detection of variants.
- The majority of genetic variants that underlie mendelian disorders disrupt protein-coding sequences.[4]
- A large number of rare nonsynonymous substitutions are predicted to be deleterious, these mutations have weak or no effects in non-coding sequences.[4]
- Splice sites also represent sequences in which there is high functional variation[4] and are therefore also included in the capture of exomes.
The exome represents an enriched portion of the genome that can be used to search for variants with large effect sizes.[4]
Target-enrichment strategies
Target-enrichment methods allow one to selectively capture genomic regions of interest from a DNA sample prior to sequencing. Several target-enrichment strategies have been developed since the original description of the direct genomic selection (DGS) method by the Lovett group in 2005.[5]
PCR

Polymerase chain reaction PCR is one of the most widely used enrichment strategies for over 20 years.[6] PCR is technology to amplify specific DNA sequences. It uses a single stranded piece of DNA as a start for DNA amplification. Uniplex PCR uses only one starting point (primer) for amplification and multiplex PCR uses multiple primers. This way multiple genes can be targeted simultaneously. This approach is known to be useful in classical Sanger sequencing because a uniplex PCR used to generate a single DNA sequence is comparable in read length to a typical amplicon. Multiplex PCR reactions which require several primers are challenging although strategies to get around this have been developed. A limitation to this method is the size of the genomic target due to workload and quantity of DNA required. The PCR based approach is highly effective, yet it is not feasible to target genomic regions that are several megabases in size due to quantity of DNA required and cost.
Molecular Inversion Probes (MIP)
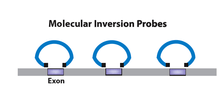
Molecular inversion probe uses probes of single stranded DNA oligonucleotides flanked by target-specific ends. The gaps between the flanking sequences are filled and ligated to form a circular DNA fragment. Probes that did not undergo reaction remain linear and are removed using exonucleases.[7][8] This is an enzymatic technique that targets the amplification of genomic regions by multiplexing based on target circularization. Accurate genotypes can be achieved from massively parallel sequencing using this method. This method is suggested to be useful for small numbers of targets in a large number of samples. Major disadvantage of this method for target enrichment is the capture uniformity as well as the cost associated with covering large target sets.[6]
Hybrid capture

Microarrays contain single-stranded oligonucleotides with sequences from the human genome to tile the region of interest fixed to the surface. Genomic DNA is sheared to form double-stranded fragments. The fragments undergo end-repair to produce blunt ends and adaptors with universal priming sequences are added. These fragments are hybridized to oligos on the microarray. Unhybridized fragments are washed away and the desired fragments are eluted. The fragments are then amplified using PCR.[7]
Roche NimbleGen was first to take the original DGS technology[5] and adapt it for next-generation sequencing. They developed the Sequence Capture Human Exome 2.1M Array to capture ~180,000 coding exons.[2] This method is both time-saving and cost-effective compared to PCR based methods. The Agilent Capture Array and the comparative genomic hybridization array are other methods that can be used for hybrid capture of target sequences. Limitations in this technique include the need for expensive hardware as well as a relatively large amount of DNA.[6]
In-solution capture
To capture genomic regions of interest using in-solution capture, a pool of custom oligonucleotides (probes) is synthesized and hybridized in solution to a fragmented genomic DNA sample. The probes (labeled with beads) selectively hybridize to the genomic regions of interest after which the beads (now including the DNA fragments of interest) can be pulled down and washed to clear excess material. The beads are then removed and the genomic fragments can be sequenced allowing for selective DNA sequencing of genomic regions (e.g., exons) of interest.
This method was developed to improve on the hybridization capture target-enrichment method. In solution capture as opposed to hybrid capture, there is an excess of probes to target regions of interest over the amount of template required.[6] The optimal target size is about 3.5 megabases and yields excellent sequence coverage of the target regions. The preferred method is dependent on several factors including; number of base pairs in the region of interest, demands for reads on target, equipment in house, etc.[9]
Sequencing
There are several sequencing platforms available including the classical Sanger sequencing. Other platforms include the Roche 454 sequencer, the Illumina Genome Analyzer II and the life technologies SOLiD & Ion Torrent all of which have been used for exome sequencing.
Significance
A study published in September 2009 discussed a proof of concept experiment to determine if it was possible to identify causal genetic variants using exome sequencing. They sequenced four individuals with Freeman-Sheldon syndrome (FSS) (OMIM 193700), a rare autosomal dominant disorder known to be caused by a mutation in the gene MYH3.[1] Eight HapMap individuals were also sequenced to remove common variants in order to identify the causal gene for FSS. After exclusion of common variants, the authors were able to identify MYH3, which confirms that exome sequencing can be used to identify causal variants of rare disorders.[1] This was the first reported study that used exome sequencing as an approach to identify an unknown causal gene for a rare mendelian disorder.
Subsequently, another group reported successful clinical diagnosis of a suspected Bartter syndrome patient of Turkish origin.[2] Bartter syndrome is a renal salt-wasting disease. Exome sequencing revealed an unexpected well-conserved recessive mutation in a gene called SLC26A3 which is associated with congenital chloride diarrhea (CLD). This molecular diagnosis of CLD was confirmed by the referring clinician. This example provided proof of concept of the use of whole-exome sequencing as a clinical tool in evaluation of patients with undiagnosed genetic illnesses. This report is regarded as the first application of next generation sequencing technology for molecular diagnosis of a patient.
A second report was conducted on exome sequencing of individuals with a mendelian disorder known as Miller syndrome (MIM#263750), a rare disorder of autosomal recessive inheritance. Two siblings and two unrelated individuals with Miller syndrome were studied. They looked at variants that have the potential to be pathogenic such as non-synonymous mutations, splice acceptor and donor sites and short coding insertions or deletions.[4] Since Miller syndrome is a rare disorder, it is expected that the causal variant has not been previously identified. Previous exome sequencing studies of common single nucleotide polymorphisms (SNPs) in public SNP databases were used to further exclude candidate genes. After exclusion of these genes, the authors found mutations in DHODH that were shared among individuals with Miller syndrome. Each individual with Miller syndrome was a compound heterozygote for the DHODH mutations which were inherited as each parent of an affected individual was found to be a carrier.[4]
This was the first time exome sequencing was shown to identify a novel gene responsible for a rare mendelian disease. This exciting finding demonstrates that exome sequencing has the potential to locate causative genes in complex diseases, which previously has not been possible due to limitations in traditional methods. Targeted capture and massively parallel sequencing represents a cost-effective, reproducible and robust strategy with high sensitivity and specificity to detect variants causing protein-coding changes in individual human genomes.
Comparison with microarray-based genotyping
There are multiple technologies available to undertake methods to identify causal genetic variants associated with disease. Each technology has its own technical, financial and throughput limitations. Microarrays, for example, require hybridization probes of a known sequence and are therefore limited by probe design and thus prevent the identification of genetic changes .[6] Massively parallel sequencing technologies, used for exome sequencing, make it now possible to identify the root of many diseases, with previously unknown causes, by screening thousands of loci at once.[10] This technology addresses the present limitations of hybridization genotyping arrays and classical sequencing.
Although exome sequencing is an expensive method relative to other technologies (e.g., hybridization-based technologies) currently available, it is an efficient strategy to identify the genetic bases that underlie rare mendelian disorders. This approach has become increasingly practical with the falling cost and increased throughput of whole genome sequencing. Even by only sequencing the exomes of individuals, a large quantity of data and sequence information is generated which requires a significant amount of data analysis. Challenges associated with the analysis of this data include changes in programs used to align and assemble sequence reads.[6] Various sequence technologies also have different error rates and generate various read-lengths which can pose challenges in comparing results from different sequencing platforms.
Limitations
Exome sequencing is only able to identify those variants found in the coding region of genes which affect protein function. It is not able to identify the structural and non-coding variants associated with the disease, which can be found using other methods such as whole genome sequencing.[1] There remains 99% of the human genome that is not covered using exome sequencing. Whole genome sequencing will eventually become a standard approach and allow us to gain a deeper understanding of genetic variation found in populations. Presently, this technique is not practical due to the high costs and time associated with sequencing large numbers of genomes. Exome sequencing allows sequencing of portions of the genome over at least 20 times as many samples compared to whole genome sequencing.[1] For translation of identified rare variants into the clinic, sample size and the ability to interpret the results to provide a clinical diagnosis indicates that with the current knowledge in genetics, exome sequencing may be the most valuable.[2]
The statistical analysis of the large quantity of data generated from sequencing approaches is a challenge. False positive and false negative findings are associated with genomic resequencing approaches and is a critical issue. A few strategies have been developed to improve the quality of exome data such as:
- Comparing the genetic variants identified between sequencing and array-based genotyping[1]
- Comparing the coding SNPs to a whole genome sequenced individual with the disorder[1]
- Comparing the coding SNPs with Sanger sequencing of HapMap individuals[1]
Rare recessive disorders would not have single nucleotide polymorphisms (SNPs) in public databases such dbSNP. More common recessive phenotypes may have disease-causing variants reported in dbSNP. For example, the most common cystic fibrosis variant has an allele frequency of about 3% in most populations. Screening out such variants might erroneously exclude such genes from consideration. Genes for recessive disorders are usually easier to identify than dominant disorders because the genes are less likely to have more than one rare nonsynonymous variant.[1] The system that screens common genetic variants relies on dbSNP which may not have accurate information about the variation of alleles. Using lists of common variation from a study exome or genome-wide sequenced individual would be more reliable. A challenge in this approach is that as the number of exomes sequenced increases, dbSNP will also increase in the number of uncommon variants. It will be necessary to develop thresholds to define the common variants that are unlikely to be associated with a disease phenotype.[10]
Genetic heterogeneity and population ethnicity are also major limitations as they may increase the number of false positive and false negative findings which will make the identification of candidate genes more difficult. Of course, it is possible to reduce the stringency of the thresholds in the presence of heterogeneity and ethnicity, however, it will reduce the power to detect variants as well.
Ethical implications
New technologies in genomics have changed the way researchers approach both basic and translational research. With approaches such as exome sequencing, it is possible to significantly enhance the data generated from individual genomes which has put forth a series of questions on how to deal with the vast amount of information. Should the individuals in these studies be allowed to have access to their sequencing information? Should this information be shared with insurance companies ? This data can lead to unexpected findings and complicate clinical utility and patient benefit. This area of genomics still remains a challenge and researchers are looking into how to address these questions.[10]
Clinical Exome Sequencing
On 29 September 2011, Ambry Genetics added the Clinical Diagnostic Exome, making them the first CLIA-certified laboratory to offer exome sequencing along with medical interpretation for clinical diagnostic purposes.[11] The company states that results from exome sequencing will allow clinicians to diagnose affected patients with conditions that have eluded traditional diagnostic approaches.
Identification of the underlying disease gene mutation(s) can have major implications for diagnostic and therapeutic approaches, can guide prediction of disease natural history, and makes it possible to test at-risk family members.[1][2][4][12][13][14] There are many factors that make exome sequencing superior to single gene analysis including the ability to identify mutations in genes that were not tested due to an atypical clinical presentation[14] or the ability to identify clinical cases where mutations from different genes contribute to the different phenotypes in the same patient.[4]
The authors of the major peer-reviewed publications on exome sequencing clearly emphasize the clinical utility. Using exome sequencing to identify the underlying mutation for a patient with Bartter Syndrome and congenital chloride diarrhea, the authors state: "We can envision a future in which such information will become part of the routine clinical evaluation of patients with suspected genetic diseases in whom the diagnosis is uncertain… We anticipate that whole-exome sequencing will make broad contributions to understanding the genes and pathways that contribute to rare and common human diseases, as well as clinical practice".[2] Bilgular's group also used exome sequencing and identified the underlying mutation for a patient with severe brain malformations, stating "[These findings]highlight the use of whole exome sequencing to identify disease loci in settings in which traditional methods have proved challenging… Our results demonstrate that this technology will be particularly valuable for gene discovery in those conditions in which mapping has been confounded by locus heterogeneity and uncertainty about the boundaries of diagnostic classification, pointing to a bright future for its broad application to medicine".[12] Likewise, whole exome sequencing was performed in a child with intractable inflammatory bowel disease. After dozens of uninformative test results, over 100 surgical procedures, and clinical consultations with physicians from around with world to little success, exome sequencing identified the underlying mutation. The mutation identification, and knowledge of the gene function, guided treatment which involved a bone marrow transplantation which cured the child of disease. In the seminal publication, the authors explain that the case "…demonstrates the power of exome sequencing to render a molecular diagnosis in an individual patient in the setting of a novel disease, after all standard diagnoses were exhausted, and illustrates how this technology can be used in a clinical setting".[13]
Direct-to-Consumer Exome Sequencing
In September 2011, the whole exome sequencing services geared toward individuals was announced by 23andMe. Individuals have had access to whole genome sequencing services for some time through companies like Illumina and Knome, but at a cost of several thousand dollars and often the services were geared toward researchers. The pilot program announced by 23andMe costs $999 and requires no physician signature, but provides only raw data without analysis.[15][16] In November 2012, DNADTC, a division of Gene by Gene started offering exomes at 80X coverage and introductory price of $695.[17] This price per DNADTC web site is currently $895. In October 2013, Beijing Genomics Institute (BGI) announced a promotion for personal whole exome sequencing at 50X coverage for $499.[18]
References
- ↑ 1.0 1.1 1.2 1.3 1.4 1.5 1.6 1.7 1.8 1.9 Ng SB, Turner EH, Robertson PD, Flygare SD, Bigham AW, Lee C, Shaffer T, Wong M, Bhattacharjee A, Eichler EE, Bamshad M, Nickerson DA, Shendure J; Turner; Robertson; Flygare; Bigham; Lee; Shaffer; Wong et al. (10 September 2009). "Targeted capture and massively parallel sequencing of 12 human exomes". Nature 461 (7261): 272–276. Bibcode:2009Natur.461..272N. doi:10.1038/nature08250. PMC 2844771. PMID 19684571.
- ↑ 2.0 2.1 2.2 2.3 2.4 2.5 2.6 Choi M, Scholl UI, Ji W, Liu T, Tikhonova IR, Zumbo P, Nayir A, Bakkaloğlu A, Ozen S, Sanjad S, Nelson-Williams C, Farhi A, Mane S, Lifton RP (10 November 2009). "Genetic diagnosis by whole exome capture and massively parallel DNA sequencing". Proc Natl Acad Sci U S A 106 (45): 19096–19101. Bibcode:2009PNAS..10619096C. doi:10.1073/pnas.0910672106. PMC 2768590. PMID 19861545.
- ↑ 3.0 3.1 3.2 Ku CS, Naidoo N, Pawitan Y. (2011). "Revisiting Mendelian disorders through exome sequencing". Human Genetics 129 (4): 351–370. doi:10.1007/s00439-011-0964-2. PMID 21331778.
- ↑ 4.0 4.1 4.2 4.3 4.4 4.5 4.6 4.7 Sarah B Ng, Kati J Buckingham, Choli Lee, Abigail W Bigham, Holly K Tabor, Karin M Dent, Chad D Huff, Paul T Shannon, Ethylin Wang Jabs, Deborah A Nickerson, Jay Shendure & Michael J Bamshad (2010). "Exome sequencing identifies the cause of a mendelian disorder". Nature Genetics 42 (1): 30–35. doi:10.1038/ng.499. PMC 2847889. PMID 19915526.
- ↑ 5.0 5.1 Stavros Basiardes, Rose Veile, Cindy Helms, Elaine R. Mardis, Anne M. Bowcock and Michael Lovett (2005). "Direct Genomic Selection". Nature Methods 1 (2): 63–69. doi:10.1038/nchembio0705-63.
- ↑ 6.0 6.1 6.2 6.3 6.4 6.5 Kahvejian A, Quackenbush J, Thompson JF (2008). "What would you do if you could sequence everything?". Nature Biotechnology 26 (10): 1125–1133. doi:10.1038/nbt1494. PMID 18846086.
- ↑ 7.0 7.1 Emily H. Turner, Sarah B. Ng, Deborah A. Nickerson, and Jay Shendure (2009). "Methods for Genomic Partitioning". Nature Genetics 10: 30–35. doi:10.1146/annurev-genom-082908-150112. PMID 19630561.
- ↑ Mertes F, Elsharawy A, Sauer S, van Helvoort JM, van der Zaag PJ, Franke A, Nilsson M, Lehrach H, Brookes AJ. (2011). "Targeted enrichment of genomic DNA regions for next-generation sequencing". Brief Funct Genomics. 10 (6): 374–386. doi:10.1093/bfgp/elr033. PMC 3245553. PMID 22121152.
- ↑ Lira Mamanova, et al.; Coffey, Alison J; Scott, Carol E; Kozarewa, Iwanka; Turner, Emily H; Kumar, Akash; Howard, Eleanor; Shendure, Jay et al. (February 2010). "Target-enrichment strategies for nextgeneration sequencing". Nature Methods 7 (2): 111–118. doi:10.1038/nmeth.1419. PMID 20111037.
- ↑ 10.0 10.1 10.2 Biesecker LG (Jan 2010). "Exome sequencing makes medical genomics a reality". Nat Genet. 42 (1): 13–14. doi:10.1038/ng0110-13. PMID 20037612.
- ↑ 'Ambry Genetics First to Offer Exome Sequencing Service for Clinical Diagnostics'
- ↑ 12.0 12.1 Bilgüvar K, Oztürk AK, Louvi A, Kwan KY, Choi M, Tatli B, Yalnizoğlu D, Tüysüz B, Cağlayan AO, Gökben S, Kaymakçalan H, Barak T, Bakircioğlu M, Yasuno K, Ho W, Sanders S, Zhu Y, Yilmaz S, Dinçer A, Johnson MH, Bronen RA, Koçer N, Per H, Mane S, Pamir MN, Yalçinkaya C, Kumandaş S, Topçu M, Ozmen M, Sestan N, Lifton RP, State MW, Günel M; Öztürk; Louvi; Kwan; Choi; Tatlı; Yalnızoğlu; Tüysüz et al. (9 September 2010). "Whole-exome sequencing identifies recessive WDR62 mutations in severe brain malformations". Nature 467 (7312): 207–210. Bibcode:2010Natur.467..207B. doi:10.1038/nature09327. PMC 3129007. PMID 20729831.
- ↑ 13.0 13.1 Worthey EA, Mayer AN, Syverson GD, Helbling D, Bonacci BB, Decker B, Serpe JM, Dasu T, Tschannen MR, Veith RL, Basehore MJ, Broeckel U, Tomita-Mitchell A, Arca MJ, Casper JT, Margolis DA, Bick DP, Hessner MJ, Routes JM, Verbsky JW, Jacob HJ, Dimmock DP. Making a definitive diagnosis: successful clinical application of whole exome sequencing in a child with intractable inflammatory bowel disease (Mar 2011). "Making a definitive diagnosis: Successful clinical application of whole exome sequencing in a child with intractable inflammatory bowel disease". Genet Med. 13 (3): 255–262. doi:10.1097/GIM.0b013e3182088158. PMID 21173700.
- ↑ 14.0 14.1 Raffan E, Hurst LA, Turki SA, Carpenter G, Scott C, Daly A, Coffey A, Bhaskar S, Howard E, Khan N, Kingston H, Palotie A, Savage DB, O'Driscoll M, Smith C, O'Rahilly S, Barroso I, Semple RK (2011). "Early Diagnosis of Werner's Syndrome Using Exome-Wide Sequencing in a Single, Atypical Patient". Front Endocrinol (Lausanne) 2 (8): 8. doi:10.3389/fendo.2011.00008. PMC 3356119. PMID 22654791.
- ↑ Herper, Matthew (27 September 2011). "The Future Is Now: 23andMe Now Offers All Your Genes For $999". Forbes. Retrieved 11 December 2011.
- ↑ "23andMe Launches Pilot Program for Direct-to-Consumer Exome Sequencing". GenomeWeb. GenomeWeb LLC. 28 September 2011. Retrieved 11 December 2011.
- ↑ Vorhaus, Dan (29 November 2012). "DNA DTC: The Return of Direct to Consumer Whole Genome Sequencing". Genomics Law Report. Retrieved 30 May 2013.
- ↑ "Ultimate Exome Promotion". BGI Americas. 18 October 2013.
External links
- WaferGen SmartChip TE Target Enrichment System
- Centrillion Exome Sequencing Service
- Roche NimbleGen Sequence Arrays
- News on Exome Sequencing
- CMS.pdf Agilent Target Enrichment System
- Guide for Patients and Families
- www.exome-seq.com