Entropy (classical thermodynamics)
| Conjugate variables of thermodynamics | |
|---|---|
| Pressure | Volume |
| (Stress) | (Strain) |
| Temperature | Entropy |
| Chemical potential | Particle number |
Entropy is a property of thermodynamical systems invented by Rudolf Clausius who named it from the Greek word τρoπή, "transformation". Later Ludwig Boltzmann described the entropy as a measure of the number of possible microscopic configurations Ω of the individual atoms and molecules of the system (microstates) which comply with the macroscopic state (macrostate) of the system. Boltzmann then went on to show that klnΩ was equal to the thermodynamic entropy. The factor k has since been known as Boltzmann's constant.
Introduction

In a thermodynamic system pressure differences, density differences, and temperature differences all tend to equalize over time. For example, consider a room containing a glass of melting ice as one system. The difference in temperature between the warm room and the cold glass of ice and water is equalized as heat from the room is transferred to the cooler ice and water mixture. Over time the temperature of the glass and its contents and the temperature of the room achieve balance. The entropy of the room has decreased. However, the entropy of the glass of ice and water has increased more than the entropy of the room has decreased. In an isolated system, such as the room and ice water taken together, the dispersal of energy from warmer to cooler regions always results in a net increase in entropy. Thus, when the system of the room and ice water system has reached temperature equilibrium, the entropy change from the initial state is at its maximum. The entropy of the thermodynamic system is a measure of how far the equalization has progressed.
There are many irreversible processes that result in an increase of the entropy. See: Entropy production. One of them is mixing of two or more different substances. The mixing is accompanied by the entropy of mixing. If the substances originally are at the same temperature and pressure, there will be no net exchange of heat or work in many important cases, such as mixing of ideal gases. The entropy increase will be entirely due to the mixing of the different substances.[1]
From a macroscopic perspective, in classical thermodynamics, the entropy is a state function of a thermodynamic system: that is, a property depending only on the current state of the system, independent of how that state came to be achieved. Entropy is a key ingredient of the Second law of thermodynamics, which has important consequences e.g. for the performance of heat engines, refrigerators, and heat pumps.
Definition
According to the Clausius equality, for a closed homogeneous system, in which only reversible processes take place,
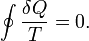
With T being the uniform temperature of the closed system and delta Q the incremental reversible transfer of heat energy into that system.
That means the line integral 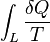 is path independent.
is path independent.
So we can define a state function S, called entropy, which satisfies
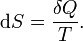
Entropy measurement
For simplicity, we examine a uniform closed system, whose thermodynamic state is determined by its temperature T and pressure P. A change in entropy can be written as
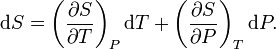
The first contribution depends on the heat capacity at constant pressure CP through

This is the result of the definition of the heat capacity by δQ = CPdT and TdS = δQ. For rewriting the second term we use one of the Maxwell relations
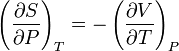
and the definition of the volumetric thermal-expansion coefficient

so that
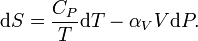
With this expression the entropy S at arbitrary P and T can be related to the entropy S0 at some reference state at P0 and T0 according to

In classical thermodynamics the entropy of the reference state can be put equal to zero at any convenient temperature and pressure. E.g., for pure substances, one can take the entropy of the solid at the melting point at 1 bar equal to zero. From a more fundamental point of view, the third law of thermodynamics suggests that there is a preference to take S = 0 at T = 0 (absolute zero) for perfectly ordered materials such as crystals.
In order to determine S(P,T) we followed a specific path in the P-T diagram: first we integrated over T at constant pressure P0, so that dP=0, and in the second integral we integrated over P at constant temperature T, so that dT=0. As the entropy is a function of state the result is independent of the path.
The above relation shows that the determination of the entropy requires knowledge of the heat capacity and the equation of state (which is the relation between P,V, and T of the substance involved). Normally these are complicated functions and numerical integration is needed. In simple cases it is possible to get analytical expressions for the entropy. E.g., in the case of an ideal gas, the heat capacity is constant and the ideal-gas law PV = nRT gives that αVV = V/T = nR/p, with n the number of moles and R the molar ideal-gas constant. So, the molar entropy of an ideal gas is given by
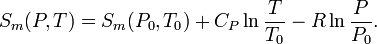
In this expression CP now is the molar heat capacity.
The entropy of inhomogeneous systems is the sum of the entropies of the various subsystems. The laws of thermodynamics hold rigorously for inhomogeneous systems even though they may be far from internal equilibrium. The only condition is that the thermodynamic parameters of the composing subsystems are (reasonably) well-defined.

Temperature-entropy diagrams
Nowadays the entropy values of important substances can be obtained via commercial software in tabular form or as diagrams. One of the most common diagrams is the temperature-entropy diagram (Ts-diagram). An example is Fig.2 which is the Ts-diagram of nitrogen.[2] It gives the melting curve and saturated liquid and vapor values together with isobars and isenthalps.
Entropy change in irreversible transformations
We now consider inhomogeneous systems in which internal transformations (processes) can take place. If we calculate the entropy S1 before and S2 after such an internal process the Second Law of Thermodynamics demands that S2 ≥ S1 where the equality sign holds if the process is reversible. The difference Si = S2 - S1 is the entropy production due to the irreversible process. The Second law demands that the entropy of an isolated system cannot decrease.
Suppose a system is thermally and mechanically isolated from the environment (isolated system). For example, consider an insulating rigid box divided by a movable partition into two volumes, each filled with gas. If the pressure of one gas is higher, it will expand by moving the partition, thus performing work on the other gas. Also, if the gases are at different temperatures, heat can flow from one gas to the other provided the partition allows heat conduction. Our above result indicates that the entropy of the system as a whole will increase during these processes. There exists a maximum amount of entropy the system may possess under the circumstances. This entropy corresponds to a state of stable equilibrium, since a transformation to any other equilibrium state would cause the entropy to decrease, which is forbidden. Once the system reaches this maximum-entropy state, no part of the system can perform work on any other part. It is in this sense that entropy is a measure of the energy in a system that cannot be used to do work.
An irreversible process degrades the performance of a thermodynamic system, designed to do work or produce cooling, and results in entropy production. The entropy generation during a reversible process is zero. Thus entropy production is a measure of the irreversibility and may be used to compare engineering processes and machines.
Thermal machines

Clausius' identification of S as a significant quantity was motivated by the study of reversible and irreversible thermodynamic transformations. A heat engine is a thermodynamic system that can undergo a sequence of transformations which ultimately return it to its original state. Such a sequence is called a cyclic process, or simply a cycle. During some transformations, the engine may exchange energy with its environment. The net result of a cycle is
- mechanical work done by the system (which can be positive or negative, the latter meaning that work is done on the engine),
- heat transferred from one part of the environment to another. In the steady state, by the conservation of energy, the net energy lost by the environment is equal to the work done by the engine.
If every transformation in the cycle is reversible, the cycle is reversible, and it can be run in reverse, so that the heat transfers occur in the opposite directions and the amount of work done switches sign.
Heat engines
Consider a heat engine working between two temperatures TH and Ta. With Ta we have ambient temperature in mind, but, in principle it may also be some other low temperature. The heat engine is in thermal contact with two heat reservoirs which are supposed to have a very large heat capacity so that their temperatures do not change significantly if heat QH is removed from the hot reservoir and Qa is added to the lower reservoir. Under normal operation TH > Ta and QH, Qa, and W are all positive.
As our thermodynamical system we take a big system which includes the engine and the two reservoirs. It is indicated in Fig.3 by the dotted rectangle. It is inhomogeneous, closed (no exchange of matter with its surroundings), and adiabatic (no exchange of heat with its surroundings). It is not isolated since per cycle a certain amount of work W is produced by the system given by the First law of thermodynamics
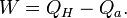
We used the fact that the engine itself is periodic, so its internal energy has not changed after one cycle. The same is true for its entropy, so the entropy increase S2 - S1 of our system after one cycle is given by the reduction of entropy of the hot source and the increase of the cold sink. The entropy increase of the total system S2 - S1 is equal to the entropy production Si due to irreversible processes in the engine so
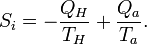
The Second law demands that Si ≥ 0. Eliminating Qa from the two relations gives
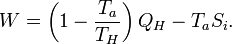
The first term is the maximum possible work for a heat engine, given by a reversible engine, as one operating along a Carnot cycle. Finally
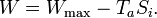
This equation tells us that the production of work is reduced by the generation of entropy. The term TaSi gives the lost work, or dissipated energy, by the machine.
Correspondingly, the amount of heat, discarded to the cold sink, is increased by the entropy generation
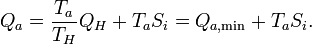
These important relations can also be obtained without the inclusion of the heat reservoirs. See the Article on entropy production.
Refrigerators
The same principle can be applied to a refrigerator working between a low temperature TL and ambient temperature. The schematic drawing is exactly the same as Fig.3 with TH replaced by TL, QH by QL, and the sign of W reversed. In this case the entropy production is

and the work, needed to extract heat QL from the cold source, is
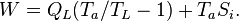
The first term is the minimum required work, which corresponds to a reversible refrigerator, so we have

i.e., the refrigerator compressor has to perform extra work to compensate for the dissipated energy due to irreversible processes which lead to entropy production.
See also
- Entropy
- Enthalpy
- Entropy production
- Fundamental thermodynamic relation
- Thermodynamic free energy
- History of entropy
- Entropy (statistical views)
References
- ↑ See, e.g., Notes for a “Conversation About Entropy” for a brief discussion of both thermodynamic and "configurational" ("positional") entropy in chemistry.
- ↑ Figure composed with data obtained with RefProp, NIST Standard Reference Database 23
Further reading
- E.A. Guggenheim Thermodynamics, an advanced treatment for chemists and physicists North-Holland Publishing Company, Amsterdam, 1959.
- C. Kittel and H. Kroemer Thermal Physics W.H. Freeman and Company, New York, 1980.
- Goldstein, Martin, and Inge F., 1993. The Refrigerator and the Universe. Harvard Univ. Press. A gentle introduction at a lower level than this entry.