Distribution (pharmacology)
Distribution in pharmacology is a branch of pharmacokinetics which describes the reversible transfer of drug from one location to another within the body.
Once a drug enters into systemic circulation by absorption or direct administration, it must be distributed into interstitial and intracellular fluids. Each organ or tissue can receive different doses of the drug and the drug can remain in the different organs or tissues for a varying amount of time.[1] The distribution of a drug between tissues is dependent on vascular permeability, regional blood flow, cardiac output and perfusion rate of the tissue and the ability of the drug to bind tissue and plasma proteins and its lipid solubility. pH partition plays a major role as well. The drug is easily distributed in highly perfused organs such as the liver, heart and kidney. It is distributed in small quantities through less perfused tissues like muscle, fat and peripheral organs. The drug can be moved from the plasma to the tissue until the equilibrium is established (for unbound drug present in plasma).
The concept of compartmentalization of an organism must be considered when discussing a drug’s distribution. This concept is used in pharmacokinetic modelling.
Factors that affect distribution
There are many factors that affect a drug’s distribution throughout an organism, but Pascuzzo[1] considers that the most important ones are the following: an organism’s physical volume, the removal rate and the degree to which a drug binds with plasma proteins and / or tissues.
Physical volume of an organism
This concept is related to multi-compartmentalization. Any drugs within an organism will act as a solute and the organism’s tissues will act as solvents. The differing specificities of different tissues will give rise to different concentrations of the drug within each group. Therefore, the chemical characteristics of a drug will determine its distribution within an organism. For example, a liposoluble drug will tend to accumulate in body fat and water-soluble drugs will tend to accumulate in extracellular fluids. The volume of distribution (VD) of a drug is a property that quantifies the extent of its distribution. It can be defined as the theoretical volume that a drug would have to occupy (if it were uniformly distributed), to provide the same concentration as it currently is in blood plasma. It can be determined from the following formula:
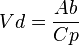 Where:
Where: 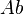 is total amount of the drug in the body and
is total amount of the drug in the body and  is the drug’s plasma concentration.
is the drug’s plasma concentration.
As the value for is equivalent to the dose of the drug that has been administered the formula shows us that there is an inversely proportional relationship between  and . That is, that the greater is the lower will be and vice versa. It therefore follows that the factors that increase will decrease . This gives an indication of the importance of knowledge relating to the drug’s plasma concentration and the factors that modify it.
and . That is, that the greater is the lower will be and vice versa. It therefore follows that the factors that increase will decrease . This gives an indication of the importance of knowledge relating to the drug’s plasma concentration and the factors that modify it.
If this formula is applied to the concepts relating to bioavailability, we can calculate the amount of drug to administer in order to obtain a required concentration of the drug in the organism ('loading dose):

This concept is of clinical interest as it is sometimes necessary to reach a certain concentration of a drug that is known to be optimal in order for it to have the required effects on the organism (as occurs if a patient is to be scanned).
Removal rate
A drug's removal rate will be determined by the proportion of the drug that is removed from circulation by each organ once the drug has been delivered to the organ by the circulating blood supply.[1] This new concept builds on earlier ideas and it depends on a number of distinct factors:
- The drugs characteristics, including its pKa.
- Redistribution through an organism’s tissues: Some drugs are distributed rapidly in some tissues until they reach equilibrium with the plasma concentration. However, other tissues with a slower rate of distribution will continue to absorb the drug from the plasma over a longer period. This will mean that the drug concentration in the first tissue will be greater than the plasma concentration and the drug will move from the tissue back into the plasma. This phenomenon will continue until the drug has reached equilibrium over the whole organism. The most sensitive tissue will therefore experience two different drug concentrations: an initial higher concentration and a later lower concentration as a consequence of tissue redistribution.
- Concentration differential between tissues.
- Exchange surface.
- Presence of natural barriers. These are obstacles to a drug's diffusion similar to those encountered during its absorption. The most interesting are:
- Capillary bed permeability, which varies between tissues.
- Blood-brain barrier: this is located between the blood plasma in the cerebral blood vessels and the brain’s extracellular space. The presence of this barrier makes it hard for a drug to reach the brain.
- Placental barrier: this prevents high concentrations of a potentially toxic drug from reaching the foetus.
Plasma protein binding
Some drugs have the capacity to bind with certain types of proteins that are carried in blood plasma. This is important as only drugs that are present in the plasma in their free form can be transported to the tissues. Drugs that are bound to plasma proteins therefore act as a reservoir of the drug within the organism and this binding reduces the drug’s final concentration in the tissues. The binding between a drug and plasma protein is rarely specific and is usually labile and reversible. The binding generally involves ionic bonds, hydrogen bonds, Van der Waals forces and, less often, covalent bonds. This means that the bond between a drug and a protein can be broken and the drug can be replaced by another substance (or another drug) and that, regardless of this, the protein binding is subject to saturation. An equilibrium also exists between the free drug in the blood plasma and that bound to proteins, meaning that the proportion of the drug bound to plasma proteins will be stable, independent of its total concentration in the plasma.
In vitro studies carried out under optimum conditions have shown that the equilibrium between a drug's plasmatic concentration and its tissue concentration is only significantly altered at binding rates to plasma proteins of greater than 90%. Above these levels the drug is "sequestered", which decreases it presence in tissues by up to 50%. This is important when considering pharmacological interactions: the tissue concentration of a drug with a plasma protein binding rate of less than 90% is not going to significantly increase if that drug is displaced from its union with a protein by another substance. On the other hand, at binding rates of greater than 95% small changes can cause important modifications in a drug’s tissue concentration. This will, in turn, increase the risk of the drug having a toxic effect on tissues.
Perhaps the most important plasma proteins are the albumins as they are present in relatively high concentrations and they readily bind to other substances. Other important proteins include the glycoproteins, the lipoproteins and to a lesser degree the globulins.
It is therefore easy to see that clinical conditions that modify the levels of plasma proteins (for example, hypoalbuminemias brought on by renal disfunction) may have an impact on the effect and toxicity of a drug that has a binding rate with plasma proteins of above 90%.
Redistribution
Highly lipid soluble drugs given by intravenous or inhalation routes are initially distributed to organs with high blood flow. Later, less vascular but more bulky tissues (such as muscle and fat) take up the drug—plasma concentration falls and the drug is withdrawn from these sites. If the site of action of the drug was in one of the highly perfused organs, redistribution results in termination of the drug action. The greater the lipid solubility of the drug, the faster its redistribution will be. For example, the anaesthetic action of thiopentone is terminated in a few minutes due to redistribution. However, when the same drug is given repeatedly or continuously over long periods, the low perfusion and high capacity sites are progressively filled up and the drug becomes longer acting.
References
See also
- Pharmacy
- Bioequivalence
- Generic drugs
- Pharmacokinetics
- Pharmacodynamics
External links
| ||||||||||||||||||||