DLVO theory
The DLVO theory is named after Derjaguin and Landau, Verwey and Overbeek.
The theory explains the aggregation of aqueous dispersions quantitatively and describes the force between charged surfaces interacting through a liquid medium.
It combines the effects of the van der Waals attraction and the electrostatic repulsion due to the so-called double layer of counterions.
The electrostatic part of the DLVO interaction is computed in the mean field approximation in the limit of low surface potentials - that is when the potential energy of an elementary charge on the surface is much smaller than the thermal energy scale, 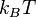 .
For two spheres of radius
.
For two spheres of radius  each having a charge
each having a charge  (expressed in units of the elementary charge)
separated by a center-to-center distance
(expressed in units of the elementary charge)
separated by a center-to-center distance  in a fluid of
dielectric constant
in a fluid of
dielectric constant 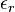 containing a concentration
containing a concentration  of monovalent ions,
the electrostatic potential takes the form of a screened-Coulomb or Yukawa repulsion,
of monovalent ions,
the electrostatic potential takes the form of a screened-Coulomb or Yukawa repulsion,
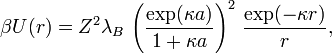
where 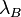 is the Bjerrum length,
is the Bjerrum length,
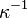 is the Debye-Hückel screening length,
which is given by
is the Debye-Hückel screening length,
which is given by  , and
, and
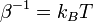 is the thermal energy scale at absolute temperature
is the thermal energy scale at absolute temperature  .
.
History
In 1923, Debye and Hückel reported the first successful theory for the distribution of charges in ionic solutions. [1] The framework of linearized Debye-Hückel theory subsequently was applied to colloidal dispersions by Levine and Dube [2] [3] who found that charged colloidal particles should experience a strong medium-range repulsion and a weaker long-range attraction. This theory did not explain the observed instability of colloidal dispersions against irreversible aggregation in solutions of high ionic strength. In 1941, Derjaguin and Landau introduced a theory for the stability of colloidal dispersions that invoked a fundamental instability driven by strong but short-ranged van der Waals attractions countered by the stabilizing influence of electrostatic repulsions. [4] Seven years later, Verwey and Overbeek independently arrived at the same result. [5] This so-called DLVO theory resolved the failure of the Levine-Dube theory to account for the dependence of colloidal dispersions' stability on the ionic strength of the electrolyte. [6]
Derivation of DLVO theory
DLVO theory is the combined effect of van der Waals and double layer force. For the derivation, different conditions must be taken into account and different equations can be obtained.[7] But some useful assumptions can effectively simplify the process, which are suitable for ordinary conditions. The simplified way to derive it is to add the two parts together.
van der Waals attraction
van der Waals force is actually the total name of dipole-dipole force, dipole-induced dipole force and dispersion forces,[8] in which dispersion forces are the most important part because they are always present. Assume that the pair potential between two atoms or small molecules is purely attractive and of the form w = -C/rn, where C is a constant for interaction energy, decided by the molecule's property and n = 6 for van der Waals attraction.[9] With another assumption of additivity, the net interaction energy between a molecule and planar surface made up of like molecules will be the sum of the interaction energy between the molecule and every molecule in the surface body.[8] So the net interaction energy for a molecule at a distance D away from the surface will therefore be

where
- w(r) is the interaction energy between the molecule and the surface
 is the number density of the surface.
is the number density of the surface.- z is the axis perpendicular with the surface and passes across the molecule. z = 0 at the point where the molecule is and z = D at the surface.
- x is the axis perpendicular with z axis, where x = 0 at the intersection.
Then the interaction energy of a large sphere of radius R and a flat surface can be calculated as
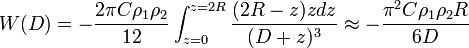
where
- W(D) is the interaction energy between the sphere and the surface.
-
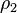 is the number density of the sphere
is the number density of the sphere
For convenience, Hamaker constant A is given as

and the equation will become
With a similar method and according to Derjaguin approximation,[10] the van der Waals interaction energy between particles with different shapes can be calculated, such as energy between
- two spheres:
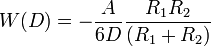
- sphere-surface:
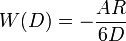
- Two surfaces:
 per unit area
per unit area
Double layer force
A surface in a liquid may be charged by dissociation of surface groups (e.g. silanol groups for glass or silica surfaces [11] ) or by adsorption of charged molecules such as polyelectrolyte from the surrounding solution. This results in the development of a wall surface potential which will attract counterions from the surrounding solution and repel co-ions. In equilibrium, the surface charge is balanced by oppositely charged counterions in solution. The region near the surface of enhanced counterion concentration is called the electrical double layer (EDL). The EDL can be approximated by a sub-division into two regions. Ions in the region closest to the charged wall surface are strongly bound to the surface. This immobile layer is called the Stern or Helmholtz layer. The region adjacent to the Stern layer is called the diffuse layer and contains loosely associated ions that are comparatively mobile. The total electrical double layer due to the formation of the counterion layers results in electrostatic screening of the wall charge and minimizes the Gibbs free energy of EDL formation.
The thickness of the diffuse electric double layer,is known as the Debye screening length  . At a distance of two Debye screening lengths the electrical potential energy is reduced to 2 percent of the value at the surface wall.
. At a distance of two Debye screening lengths the electrical potential energy is reduced to 2 percent of the value at the surface wall.
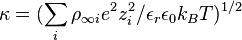
with unit of m−1 where
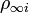 is the number density of ion i in the bulk solution.
is the number density of ion i in the bulk solution.- z is the valency of the ion. For example, H+ has a valency of +1 and Ca2+ has a valency of +2.
 is the vacuum permittivity, is the relative static permittivity.
is the vacuum permittivity, is the relative static permittivity.- kB is the Boltzmann constant.
The repulsive free energy per unit area between two planar surfaces is shown as
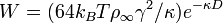
where
 is the reduced surface potential
is the reduced surface potential
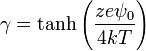
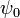 is the potential on the surface.
is the potential on the surface.
The interaction free energy between two spheres of radius R is
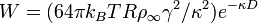
Combining the van der Waals interaction energy and the double layer interaction energy, the interaction between two particles or two surfaces in a liquid can be expressed as:

where W(D)R is the repulsive interaction energy due to electric repulsion and W(D)A is the attractive interaction energy due to van der Waals interaction.
Application of DLVO theory
Since the 1940s, the DLVO theory has been used to explain phenomena found in colloidal science, adsorption and many other fields. Due to the more recent popularity of nanoparticle research, DLVO theory has become even more popular because it can be used to explain behavior of both material nanoparticles such as fullerene particles and microorganisms.
Shortcomings of the DLVO theory
Additional forces beyond the DLVO construct have been reported to also play a major role in determining colloid stability. [13] DLVO theory is not effective in describing ordering processes such as the evolution of colloidal crystals in dilute dispersions with low salt concentrations. It also can not explain the relation between the formation of colloidal crystals and salt concentrations. [14]
References
- ↑ Debye, P.; Hückel, E. (1923), "The theory of electrolytes. I. Lowering of freezing point and related phenomena", Physikalische Zeitschrift 24: 185–206.
- ↑ Levine, S. (1939), "Problems of stability in hydrophobic colloidal solutions I. On the interaction of two colloidal metallic particles. General discussion and applications", Proceedings of the Royal Society of London A 170 (145): 165.
- ↑ Levine, S.; Dube, G. P. (1940), "Interaction between two hydrophobic colloidal particles, using the approximate Debye-Huckel theory. I. General properties", Transactions of the Faraday Society 35: 1125–1141.
- ↑ Derjaguin, B.; Landau, L. (1941), "Theory of the stability of strongly charged lyophobic sols and of the adhesion of strongly charged particles in solutions of electrolytes", Acta Physico Chemica URSS 14: 633.
- ↑ Verwey, E. J. W.; Overbeek, J. Th. G. (1948), Theory of the stability of lyophobic colloids, Amsterdam: Elsevier.
- ↑ Russel, W. B.; Saville, D. A.; Schowalter, W. R. (1989), Colloidal Dispersions, New York: Cambridge University Press.
- ↑ M Elimelech, J Gregory, X Jia, R A Williams, Particle Deposition and Aggregation Measurement: Modelling and Simulation (Boston:1995).
- ↑ 8.0 8.1 Jacob N. Israelacvili, Intermolecular and Surface Forces (London 2007).
- ↑ London, F. (1937), Trans Faraday Soc, 33, 8-26.
- ↑ Derjaguin B. V. (1934)Kolloid Zeits 69, 155-164.
- ↑ Behrens, S. H. and Grier, D. G., "The charge on glass and silica surfaces," Journal of Chemical Physics 115, 6716-6721 (2001)
- ↑ Bhattacharjee, S.; Elimelech, M.; Borkovec, Michal (1998), "DLVO interaction between colloidal particles: Beyond Derjaguins approximation", Croatica Chimca Acta 71: 883–903.
- ↑ Grasso, D., Subramaniam, K., Butkus,M., K Strevett, Bergendahl, J. "A review of non-DLVO interactions in environmental colloidal systems," Reviews in Environmental Science and Biotechnology 1 (1), 17-38
- ↑ N. Ise and I. S. Sogami, Structure Formation in Solution: Ionic Polymers and Colloidal Particles, (Springer, New York, 2005).