Cre-Lox recombination
In the field of genetics, Cre-Lox recombination is known as a site-specific recombinase technology, and is widely used to carry out deletions, insertions, translocations and inversions at specific sites in the DNA of cells. It allows the DNA modification to be targeted to a specific cell type or be triggered by a specific external stimulus. It is implemented both in eukaryotic and prokaryotic systems.
The system consists of a single enzyme, Cre recombinase, that recombines a pair of short target sequences called the Lox sequences. This system can be implemented without inserting any extra supporting proteins or sequences. The Cre enzyme and the original Lox site called the LoxP sequence are derived from bacteriophage P1.
Placing Lox sequences appropriately allows genes to be activated, repressed, or exchanged for other genes. At a DNA level many types of manipulations can be carried out. The activity of the Cre enzyme can be controlled so that it is expressed in a particular cell type or triggered by an external stimulus like a chemical signal or a heat shock. These targeted DNA changes are useful in cell lineage tracing and when mutants are lethal if expressed globally.
The Cre-Lox system is very similar in action and in usage to the FLP-FRT recombination system.[1]
History
Cre-Lox recombination is a special type of site-specific recombination developed by Dr. Brian Sauer initially for use in activating gene expression in mammalian cell lines and transgenic mice (DuPont).[2][3] Subsequently, workers in the laboratory of Pr Nathalie JANEL showed that Cre-Lox recombination could be used to delete loxP-flanked chromosomal DNA sequences at high efficiency in selected cell types of transgenic animals, suggesting this approach as a means to define gene function in specific cell types, indelibly mark progenitors in cell fate determination studies, induce specific chromosomal rearrangements for biological and disease modeling, and determine the roles of early genetic lesions in disease (and phenotype) maintenance.[4] Shortly thereafter, the laboratory of Dr. Klaus Rajewsky reported the production of pluripotent embryonic stem cells bearing a targeted loxP-flanked (floxed) DNA polymerase gene.[5] Combining these advances in collaboration, the laboratories of Drs. Marth and Rajewsky showed in 1994 that Cre-lox recombination could be used for conditional gene targeting in vivo.[6] This technique continues to be used by hundreds of researchers and laboratories around the world as an essential procedure to discover gene function in normal and disease biology, resulting in numerous important discoveries that would otherwise have not been possible.
Overview
Cre-Lox recombination involves the targeting of a specific sequence of DNA and splicing it with the help of an enzyme called Cre recombinase. Because systemic inactivation of many genes further cause embryonic lethality, Cre-Lox recombination is commonly used to circumvent this problem. In addition, Cre–Lox recombination provides the best experimental control that presently exists in transgenic animal modeling to link genotypes (alterations in genomic DNA) to the biological outcomes (phenotypes).
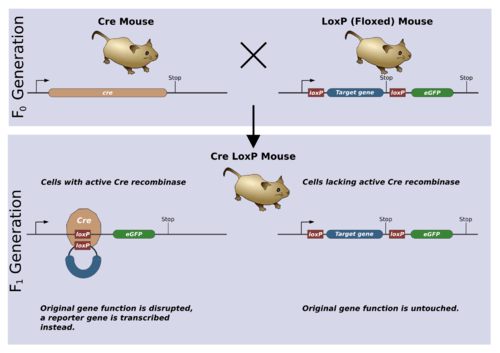
The Cre-lox system is used as a genetic tool to control site specific recombination events in genomic DNA. This system has allowed researchers to manipulate a variety of genetically modified organisms to control gene expression, delete undesired DNA sequences and modify chromosome architecture.
The Cre protein is a site-specific DNA recombinase, that is, it can catalyse the recombination of DNA between specific sites in a DNA molecule. These sites, known as loxP sequences, contain specific binding sites for Cre that surround a directional core sequence where recombination can occur.

When cells that have loxP sites in their genome express Cre, a recombination event can occur between the loxP sites. Cre recombinase proteins binds to the first and last 13 bp regions of a lox site forming a dimer. This dimer then binds to a dimer on another lox site to form a tetramer. Lox sites are directional and the two sites joined by the tetramer are parallel in orientation. The double stranded DNA is cut at both loxP sites by the Cre protein. The strands are then rejoined with DNA ligase in a quick and efficient process. The result of recombination depends on the orientation of the loxP sites. For two lox sites on the same chromosome arm, inverted loxP sites will cause an inversion of the intervening DNA, while a direct repeat of loxP sites will cause a deletion event. If loxP sites are on different chromosomes it is possible for translocation events to be catalysed by Cre induced recombination. Two plasmids can be joined using the variant lox sites 71 and 66.[7]
Cre recombinase
The Cre protein (encoded by the locus originally named as "Causes recombination", with "Cyclization recombinase" being found in some references)[8][9] consists of 4 subunits and two domains: The larger carboxyl (C-terminal) domain, and smaller amino (N-terminal) domain. The total protein has 343 amino acids. The C domain is similar in structure to the domain in the Integrase family of enzymes isolated from lambda phage. This is also the catalytic site of the enzyme.
Lox P site
Lox P (locus of X-over P1) is a site on the bacteriophage P1 consisting of 34 bp. The site includes an asymmetric 8 bp sequence, variable except for the middle two bases, in between two sets of palindromic, 13 bp sequences. The exact sequence is given below; 'N' indicates bases which may vary.
| 13bp | 8bp | 13bp |
| ATAACTTCGTATA - | NNNTANNN | -TATACGAAGTTAT |
Holliday junctions and homologous recombination
During genetic recombination, a Holliday junction is formed between the two strands of DNA and a double-stranded break in a DNA molecule leaves a 3’OH end exposed. This reaction is aided with the endonuclease activity of an enzyme. 5’ Phosphate ends are usually the substrates for this reaction, thus extended 3’ regions remain. This 3’ OH group is highly unstable, and the strand on which it is present must find its complement. Since Homologous Recombination occurs after DNA replication, two strands of DNA are available, and, thus, the 3’ OH group must pair with its complement, and it does so, with an intact strand on the other duplex. Now, one point of crossover has occurred, which is what is called a Holliday Intermediate.
The 3’OH end is elongated (that is, bases are added) with the help of DNA Polymerase. The pairing of opposite strands is what constitutes the crossing-over or Recombination event, which is common to all living organisms, since the genetic material on one strand of one duplex has paired with one strand of another duplex, and has been elongated by DNA polymerase. Further cleavage of Holliday Intermediates results in formation of Hybrid DNA.
This further cleavage or ‘resolvation’ is done by a special group of enzymes called Resolvases. RuvC is just one of these Resolvases that have been isolated in bacteria and yeast.
For many years, it was thought that when the Holliday junction intermediate was formed, the branch point of the junction (where the strands cross over) would be located at the first cleavage site. Migration of the branch point to the second cleavage site would then somehow trigger the second half of the pathway. This model provided convenient explanation for the strict requirement for homology between recombining sites, since branch migration would stall at a mismatch and would not allow the second strand exchange to occur. In more recent years, however, this view has been challenged, and most of the current models for Int, Xer, and Flp recombination involve only limited branch migration 1–3 base pairs) of the Holliday intermediate, coupled to an isomerisation event that is responsible for switching the strand cleavage specificity.
Site-specific recombination
Site-specific recombination (SSR) involves specific sites for the catalysing action of special enzymes called recombinases. Cre, or cyclic recombinase, is one such enzyme. Site-specific recombination is, thus, the enzyme-mediated cleavage and ligation of two defined deoxynucleotide sequences.
A number of conservative site-specific recombination systems have been described in both prokaryotic and eukaryotic organisms. In general, these systems use one or more proteins and act on unique asymmetric DNA sequences. The products of the recombination event depend on the relative orientation of these asymmetric sequences. Many other proteins apart from the Recombinase are involved in regulating the reaction. During site-specific DNA recombination, which brings about genetic rearrangement in processes such as viral integration and excision and chromosomal segregation, these recombinase enzymes recognize specific DNA sequences and catalyse the reciprocal exchange of DNA strands between these sites.
Mechanism of action
Initiation of site-specific recombination begins with the binding of recombination proteins to their respective DNA targets. A separate recombinase recognizes and binds to each of two recombination sites on two different DNA molecules or within the same DNA. At the given specific site on the DNA, the hydroxyl group of the tyrosine attacks a phosphate group in the DNA using a direct transesterification mechanism linking the recombinase protein to the DNA via a phospho-tyrosine linkage. This conserves the energy of the phosphodiester bond, allowing the reaction to be reversed without the involvement of a high-energy cofactor.
Cleavage on the other strand also causes a phospho-tyrosine bond between DNA and the enzyme. At both the DNA duplexes, the bonding of the phosphate group to tyrosine residues leave a 3’ OH group free. In fact, the enzyme-DNA complex is an intermediate stage, which is followed by the ligation of the 3’ OH group of one DNA strand to the 5’ phosphate group of the other DNA strand, which is covalently bonded to the tyrosine residue; that is, the covalent linkage between 5’ end and tyrosine residue is broken. This reaction synthesizes the Holliday Junction discussed earlier.
In this fashion, opposite DNA strands are joined together. Subsequent cleavage and rejoining cause DNA strands to exchange their segments. Protein-Protein interactions drive and direct strand exchange. Energy is not compromised, since the Protein-DNA linkage makes up for the loss of the Phosphodiester bond, which occurred during cleavage.
Site-specific Recombination is also an important process that viruses, such as bacteriophages, adopt to integrate their genetic material into the infected host. The virus, called a prophage in such a state, accomplishes this via integration and excision. The points where the integration and excision reactions occur are called the attachment (att) sites. An attP site on the Phage exchanges segments with an attB site on the Bacterial DNA. Thus, these are site-specific, occurring only at the respective att sites. The integrase class of enzymes catalyse this particular reaction.
The natural function of the Cre-lox system
The P1 phage is a temperate phage that causes either a lysogenic or lytic cycle when it infects a bacterium. In its lytic state, once its viral genome is injected into the host cell, viral proteins are produced, virions are assembled, and the host cell is lysed to release the phages, continuing the cycle. In the lysogenic cycle the phage genome replicates with the rest of the bacterial genome and is transmitted to daughter cells at each subsequent cell division. It can transition to the lytic cycle by a later event such as UV radiation or starvation.
Phages like the lambda phage use their site specific recombinases to integrate their DNA into the host genome during lysogeny. P1 phage DNA on the other hand, exists as a plasmid in the host. The Cre-lox system serves several functions in the phage: it circularizes the phage DNA into a plasmid, separates interlinked plasmid rings so they are passed to both daughter bacteria equally and may help maintain copy numbers through an alternative means of replication.[10]
The P1 phage DNA when released into the host from the virion is in the form of a linear double stranded DNA molecule. The Cre enzyme targets loxP sites at the ends of this molecule and cyclises the genome. This can also take place in the absence of the Cre lox system[11] with the help of other bacterial and viral proteins. The P1 plasmid is relatively large (~90Kbp) and hence exists in a low copy number - usually one per cell. If the two daughter plasmids get interlinked one of the daughter cells of the host will lose the plasmid. The Cre-lox recombination system prevents these situations by unlinking the rings of DNA by carrying out two recombination events (linked rings -> single fused ring -> two unlinked rings). It is also proposed that rolling circle replication followed by recombination will allow the plasmid to increase its copy number when certain regulators (repA) are limiting.[10]
Implementation of multiple loxP site pairs
Multiple variants of loxP,[12] in particular lox2272 and loxN, have been used by researchers with the combination of different Cre actions (transient or constitutive) to create a "Brainbow" system that allows multi-colouring of mice's brain with four fluorescent proteins.
References
- ↑ Turan, S.; Galla, M.; Ernst, E.; Qiao, J.; Voelkel, C.; Schiedlmeier, B.; Zehe, C.; Bode, J. (2011). "Recombinase-mediated cassette exchange (RMCE): traditional concepts and current challenges". J. Mol. Biol. 407 (2): 193–221. doi:10.1016/j.jmb.2011.01.004. PMID 21241707.
- ↑ Sauer, B. (1987). "Functional expression of the Cre-Lox site-specific recombination system in the yeast Saccharomyces cerevisiae". Mol Cell Biol 7: 2087–2096.
- ↑ Sauer, B.; Henderson, N. (1988). "Site-specific DNA recombination in mammalian cells by the Cre recombinase of bacteriophage P1". Proc. Natl. Acad. Sci. USA 85: 5166–5170.
- ↑ Orban, P.C., Chui, D., and Marth, J.D. (1992) "Tissue– and site–specific recombination in transgenic mice." "Proc. Natl. Acad. Sci. USA" "89": 6861–6865
- ↑ Gu, H., Zou, Y.R., and Rajewsky, K. (1993) "Independent control of immunoglobulin switch recombination at individual switch regions evidenced through Cre–loxP–mediated gene targeting." "Cell" "73": 1155-1564
- ↑ Gu, H., Marth, J.D., Orban, P.C., Mossman, H., and Rajewsky, K. (1994) "Deletion of the DNA polymerase beta gene in T cells using tissue-specific gene targeting. "Science" "265": 103-106.
- ↑ Hastie and Pruitt "Yeast two-hybrid interaction partner screening through in vivo Cre-mediated Binary Interaction Tag generation" Nucleic Acids Research (2007) 35[21], e141
- ↑ http://www.ncbi.nlm.nih.gov/protein/AAV84941.1
- ↑ Sternberg and Hamilton, J.Mol.Biol. (1981) 150, 467-486, pp 468
- ↑ 10.0 10.1 Łobocka, Małgorzata B.; Debra J. Rose, Guy Plunkett, Marek Rusin, Arkadiusz Samojedny, Hansjörg Lehnherr, Michael B. Yarmolinsky, Frederick R. Blattner (November 2004). "Genome of Bacteriophage P1". Journal of Bacteriology 186 (21): 7032–7068. doi:10.1128/JB.186.21.7032-7068.2004. ISSN 0021-9193. PMC 523184. PMID 15489417.
- ↑ Sternberg, N; R Hoess (1983). "The Molecular Genetics of Bacteriophage P1". Annual Review of Genetics 17 (1): 123–154. doi:10.1146/annurev.ge.17.120183.001011. Retrieved 2012-03-26.
- ↑ Editor's Summary (1 November 2007) Over the brainbow. Nature. Accessed 15 March 2010.