Conformational entropy
Conformational entropy is the entropy associated with the physical arrangement of a polymer chain that assumes a compact or globular state in solution. The concept is most commonly applied to biological macromolecules such as proteins and RNA, but can also be used for polysaccharides and other polymeric organic compounds. To calculate the conformational entropy, the possible conformations assumed by the polymer may first be discretized into a finite number of states, usually characterized by unique combinations of certain structural parameters, each of which has been assigned an energy level. In proteins, backbone dihedral angles and side chain rotamers are commonly used as descriptors, and in RNA the base pairing pattern is used. These characteristics are used to define the degrees of freedom (in the statistical mechanics sense of a possible "microstate"). The conformational entropy associated with a particular conformation is then dependent on the probability associated with the occupancy of that state, as determined by the sum of the energies associated with the value of each parameter assumed in the state.
The entropy of heterogeneous random coil or denatured proteins is significantly higher than that of the folded native state tertiary structure. In particular, the conformational entropy of the amino acid side chains in a protein is thought to be a major contributor to the energetic stabilization of the denatured state and thus a barrier to protein folding.[1] However, a recent study has shown that side-chain conformational entropy can stabilize native structures among alternative compact structures.[2] The conformational entropy of RNA and proteins can be estimated; for example, empirical methods to estimate the loss of conformational entropy in a particular side chain on incorporation into a folded protein can roughly predict the effects of particular point mutations in a protein. Side-chain conformational entropies can be defined as Boltzmann sampling over all possible rotameric states:[3]

where  is the gas constant and
is the gas constant and 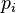 is the probability of a residue being in rotamer
is the probability of a residue being in rotamer  .[3]
.[3]
The limited conformational range of proline residues lowers the conformational entropy of the denatured state and thus increases the energy difference between the denatured and native states. A correlation has been observed between the thermostability of a protein and its proline residue content.[4]
References
- ↑ Doig AJ, Sternberg MJE. (1995). Side-chain conformational entropy in protein folding. Protein Science 4:2247-51.
- ↑ Zhang J, Liu JS (2006) On Side-Chain Conformational Entropy of Proteins. PLoS Comput Biol 2(12): e168. doi:10.1371/journal.pcbi.0020168
- ↑ 3.0 3.1 Pickett SD, Sternberg MJ. (1993). Empirical scale of side-chain conformational entropy in protein folding. J Mol Biol 231(3):825-39.
- ↑ Watanabe K., Masuda T., Ohashi H., Mihara H. & Suzuki Y. Multiple proline substitutions cumulatively thermostabilize Bacillus cereus ATCC7064 oligo-1,6-glucosidase. Irrefragable proof supporting the proline rule. Eur J Biochem 226,277-83 (1994).