Chromatin remodeling
Chromatin remodeling is the dynamic modification of chromatin architecture to allow access of condensed genomic DNA to the regulatory transcription machinery proteins, and thereby control gene expression. Such remodeling is principally carried out by 1) covalent histone modifications by specific enzymes, i.e., histone acetyltransferases (HATs), deacetylases, methyltransferases, and kinases, and 2) ATP-dependent chromatin remodeling complexes which either move, eject or restructure nucleosomes.[1] Besides actively regulating gene expression, dynamic remodeling of chromatin imparts an epigenetic regulatory role in several key biological processes, e.g., DNA replication and repair; apoptosis; chromosome segregation as well as development and pluripotency. Aberrations in chromatin remodeling proteins are found to be associated with human diseases, including cancer. Targeting chromatin remodeling pathways is currently evolving as a major therapeutic strategy in the treatment of several cancers.
Overview
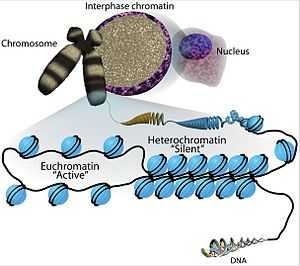
The transcriptional regulation of the genome is controlled primarily at the preinitiation stage by binding of the core transcriptional machinery proteins (namely, RNA polymerase, transcription factors, and activators and repressors) to the core promoter sequence on the coding region of the DNA. However, DNA is tightly packaged in the nucleus with the help of packaging proteins, chiefly histone proteins to form repeating units of nucleosomes which further bundle together to form condensed chromatin structure. Such condensed structure occludes many DNA regulatory regions, not allowing them to interact with transcriptional machinery proteins and regulate gene expression. To overcome this issue and allow dynamic access to condensed DNA, a process known as chromatin remodeling alters nucleosome architecture to expose or hide regions of DNA for transcriptional regulation.
By definition, chromatin remodeling is the enzyme-assisted process to facilitate access of nucleosomal DNA by remodeling the structure, composition and positioning of nucleosomes.
Classification
Access to nucleosomal DNA is governed by two major classes of protein complexes:
- Covalent histone-modifying complexes.
- ATP-dependent chromatin remodeling complexes.
Covalent histone-modifying complexes
Specific protein complexes, known as histone-modifying complexes catalyze addition or removal of various chemical elements on histones. These enzymatic modifications include acetylation, methylation, phosphorylation, and ubiquitination and primarily occur at N-terminal histone tails. Such modifications affect the binding affinity between histones and DNA, and thus loosening and tightening the condensed DNA wrapped around histones, e.g., Methylation of specific lysine residues in H3 and H4 causes further condensation of DNA around histones, and thereby preventing binding of transcription factors to the DNA leading to gene repression. On contrary, histone acetylation relaxes chromatin condensation and exposes DNA for TF binding, leading to increase gene expression.[2]
Known modifications
Well characterized modifications to histones include:[3]
Both lysine and arginine residues are known to be methylated. Methylated lysines are the best understood marks of the histone code, as specific methylated lysine match well with gene expression states. Methylation of lysines H3K4 and H3K36 is correlated with transcriptional activation while demethylation of H3K4 is correlated with silencing of the genomic region. Methylation of lysines H3K9 and H3K27 is correlated with transcriptional repression.[4] Particularly, H3K9me3 is highly correlated with constitutive heterochromatin.[5]
- Acetylation - by HAT (histone acetyl transferase); deacetylation - by HDAC (histone deacetylase)
Acetylation tends to define the ‘openness’ of chromatin as acetylated histones cannot pack as well together as deacetylated histones.
- Phosphorylation
- Ubiquitination
However there are many more histone modifications, and sensitive mass spectrometry approaches have recently greatly expanded the catalog.[6]
Histone code hypothesis
The histone code is a hypothesis that the transcription of genetic information encoded in DNA is in part regulated by chemical modifications to histone proteins, primarily on their unstructured ends. Together with similar modifications such as DNA methylation it is part of the epigenetic code.
Cumulative evidences suggest that such code is 1) written by specific enzymes which can (for example) methylate or acetylate DNA ('writers'), 2) removed by other enzymes having demethylase or deacetylase activity ('erasers'), and finally 3) readily identified by proteins (‘readers’) that are recruited to such histone modifications and bind via specific domains, e.g., bromodomain, chromodomain. These triple action of ‘writing’, ‘reading’ and ‘erasing’ establish the favorable local environment for transcriptional regulation, DNA-damage repair, etc.[7]
The critical concept of the histone code hypothesis is that the histone modifications serve to recruit other proteins by specific recognition of the modified histone via protein domains specialized for such purposes, rather than through simply stabilizing or destabilizing the interaction between histone and the underlying DNA. These recruited proteins then act to alter chromatin structure actively or to promote transcription.
A very basic summary of the histone code for gene expression status is given below (histone nomenclature is described here):
| Type of modification |
Histone | ||||||
|---|---|---|---|---|---|---|---|
| H3K4 | H3K9 | H3K14 | H3K27 | H3K79 | H4K20 | H2BK5 | |
| mono-methylation | activation[8] | activation[9] | activation[9] | activation[9][10] | activation[9] | activation[9] | |
| di-methylation | repression[4] | repression[4] | activation[10] | ||||
| tri-methylation | activation[11] | repression[9] | repression[9] | activation,[10] repression[9] |
repression[4] | ||
| acetylation | activation[11] | activation[11] | |||||
ATP-dependent chromatin remodeling

ATP-dependent chromatin-remodeling complexes regulate gene expression by either moving, ejecting or restructuring nucleosomes. These protein complexes have a common ATPase domain and energy from the hydrolysis of ATP allows these remodeling complexes to reposition (slide, twist or loop) nucleosomes along the DNA, expel histones away from DNA or facilitate exchange of histone variants, and thus creating nucleosome-free regions of DNA for gene activation.[12] Also, several remodelers have DNA-translocation activity to carry out specific remodeling tasks.[13]
Known chromatin remodeling complexes
There are at least five families of chromatin remodelers in eukaryotes : SWI/SNF, ISWI, NuRD/Mi-2/CHD, INO80 and SWR1 with first two remodelers being very well studied so far, especially in the yeast model. Although all of remodelers share common ATPase domain, their functions are specific based on several biological processes (DNA repair, apoptosis, etc.). This is due to the fact that each remodeler complex has unique protein domains (Helicase, bromodomain, etc.) in their catalytic ATPase region and also has different recruited subunits.
Specific functions
- Several in-vitro experiments suggest that ISWI remodelers organize nucleosome into proper bundle form and create equal spacing between nucleosomes, whereas SWI/SNF remodelers disorder nucleosomes.
- The ISWI-family remodelers have been shown to play central roles in chromatin assembly after DNA replication and maintenance of higher-order chromatin structures.
- INO80 and SWI/SNF-family remodelers participate in DNA double-strand break (DSB) repair and nucleotide-excision repair (NER) and thereby plays crucial role in TP53 mediated DNA-damage response.
- NuRD/Mi-2/CHD remodeling complexes primarily mediate transcriptional repression in the nucleus as well as required for the maintenance of pluripotency of embryonic stem cells.[12]
Significance
In normal biological processes
Chromatin remodeling plays a central role in the regulation of gene expression by providing the transcription machinery with dynamic access to an otherwise tightly packaged genome. Further, nucleosome movement by chromatin remodelers is essential to several important biological processes, including chromosome assembly and segregation, DNA replication and repair, embryonic development and pluripotency, and cell-cycle progression. Deregulation of chromatin remodeling causes loss of transcriptional regulation at these critical check-points required for proper cellular functions, and thus causes various disease syndromes, including cancer.
Cancer
Chromatin remodeling provides fine-tuning at crucial cell growth and division steps, like cell-cycle progression, DNA repair and chromosome segregation, and therefore exerts tumor-suppressor function. Mutations in such chromatin remodelers and deregulated covalent histone modifications potentially favor self-sufficiency in cell growth and escape from growth-regulatory cell signals - two important hallmarks of cancer.[14]
- Inactivating mutations in SMARCB1, formerly known as hSNF5/INI1 and a component of the human SWI/SNF remodeling complex have been found in large number of rhabdoid tumors, commonly affecting pediatric population.[15] Similar mutations are also present in other childhood cancers, such as choroid plexus carcinoma, medulloblastoma and in some acute leukemias. Further, mouse knock-out studies strongly support SMARCB1 as a tumor suppressor protein. Since the original observation of SMARCB1 mutations in rhabdoid tumors, several more subunits of the human SWI/SNF chromatin remodeling complex have been found mutated in a wide range of neoplasms.[16]
- PML-RAR fusion protein in acute myeloid leukemia recruits histone deacetylases. This leads to repression of gene responsible for myelocytes to differentiate, leading to leukemia.
- Tumor suppressor Rb protein functions by the recruitment of the human homologs of the SWI/SNF enzymes BRG1, histone deacetylase and DNA methyltransferase. Mutations in BRG1 are reported in several cancers causing loss of tumor suppressor action of Rb.[17]
- Recent reports indicate DNA hypermethylation in the promoter region of major tumor suppressor genes in several cancers. Although few mutations are reported in histone methyltransferases yet, correlation of DNA hypermethylation and histone H3 lysine-9 methylation has been reported in several cancers, mainly in colorectal and breast cancers.
- Mutations in Histone Acetyl Transferases (HAT) p300 (missense and truncating type) are most commonly reported in colorectal, pancreatic, breast and gastric carcinomas. Loss of heterozygosity in coding region of p300 (chromosome 22q13) is present in large number of glioblastomas.
- Further, HATs have diverse role as transcription factors beside having histone acetylase activity, e.g., HAT subunit, hADA3 may act as an adaptor protein linking transcription factors with other HAT complexes. In the absence of hADA3, TP53 transcriptional activity is significantly reduced, suggesting role of hADA3 in activating TP53 function in response to DNA-damage.
- Similarly, TRRAP, the human homolog to yeast Tra1, has been shown to directly interact with c-Myc and E2F1 - known oncoproteins.
Cancer genomics
Rapid advance in cancer genomics and high-throughput ChIP-chip, ChIP-Seq and Bisulfite sequencing methods are providing more insight into role of chromatin remodeling in transcriptional regulation and role in cancer.
Therapeutic intervention
Epigenetic instability caused by deregulation in chromatin remodeling is studied in several cancers, including breast cancer, colorectal cancer, pancreatic cancer. Such instability largely cause widespread silencing of genes with primary impact on tumor-suppressor genes. Hence, strategies are now being tried to overcome epigenetic silencing with synergistic combination of HDAC inhibitors or HDI and DNA-demethylating agents. HDIs are primarily used as adjunct therapy in several cancer types.[18][19] HDAC inhibitors can induce p21 (WAF1) expression, a regulator of p53's tumor suppressoractivity. HDACs are involved in the pathway by which the retinoblastoma protein (pRb) suppresses cell proliferation.[20] Estrogens well-established as a mitogenic factorimplicated in the tumorigenesis and progression of breast cancer via its binding to the estrogen receptor alpha (ERα). Recent data indicate that chromatin inactivation mediated by HDAC and DNA methylation is a critical component of ERα silencing in human breast cancer cells.[21]
- Approved usage:
- Vorinostat was licenced by the U.S. FDA in October 2006 for the treatment of cutaneous T cell lymphoma (CTCL).
- Romidepsin (trade name Istodax) was licenced by the US FDA in Nov 2009 for cutaneous T-cell lymphoma (CTCL).
- Phase III Clinical trials:
- Panobinostat (LBH589) is in clinical trials for various cancers including a phase III trial for cutaneous T cell lymphoma (CTCL).
- Valproic acid (as Mg valproate) in phase III trials for cervical cancer and ovarian cancer.
Started pivotal phase II clinical trials
- Belinostat (PXD101) has had a phase II trial for relapsed ovarian cancer, and reported good results for T cell lymphoma.
- Click here for more
Current front-runner candidates for new drug targets are Histone Lysine Methyltransferases (KMT) and Protein Arginine Methyltransferases (PRMT).[22]
Other disease syndromes
- ATRX-syndrome (α-thalassemia X-linked mental retardation) and α-thalassemia myelodysplasia syndrome are caused by mutations in ATRX, a SNF2-related ATPase with a PHD.
- CHARGEsyndrome, an autosomal dominant disorder, has been linked recently to haploinsufficiency of CHD7, which encodes the CHD family ATPase CHD7.[23]
See also
- Epigenetics
- Histone
- Nucleosomes
- Chromatin
- Histone acetyltransferase
- Transcription factors
References
- ↑ Teif VB, Rippe K. (2009). "Predicting nucleosome positions on the DNA: combining intrinsic sequence preferences and remodeler activities.". Nucleic Acids Res. 37 (17): 5641–55. doi:10.1093/nar/gkp610. PMC 2761276. PMID 19625488.
- ↑ Wang GG, Allis CD, Chi P. (2007). "Chromatin remodeling and cancer, Part II: ATP-dependent chromatin remodeling.". Trends Mol Med. 13 (9): 363–72. doi:10.1016/j.molmed.2007.07.003. PMID 17822958.
- ↑ Strahl B, Allis C (2000). "The language of covalent histone modifications". Nature 403 (6765): 41–5. doi:10.1038/47412. PMID 10638745.
- ↑ 4.0 4.1 4.2 4.3 Rosenfeld, Jeffrey A; Wang, Zhibin; Schones, Dustin; Zhao, Keji; DeSalle, Rob; Zhang, Michael Q (31 March 2009). "Determination of enriched histone modifications in non-genic portions of the human genome.". BMC Genomics 10: 143. doi:10.1186/1471-2164-10-143. PMC 2667539. PMID 19335899. Unknown parameter
|unused_data=ignored (help) - ↑ Hublitz, Philip; Albert, Mareike; Peters, Antoine (28 April 2009). "Mechanisms of Transcriptional Repression by Histone Lysine Methylation". The International Journal of Developmental Biology (Basel) 10 (1387): 335–354. ISSN 1696-3547.
- ↑ Tan M, Luo H, Lee S, Jin F, Yang JS, Montellier E et al. (2011). "Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification.". Cell 146 (6): 1016–28. doi:10.1016/j.cell.2011.08.008. PMC 3176443. PMID 21925322.
- ↑ Jenuwein T, Allis C (2001). "Translating the histone code". Science 293 (5532): 1074–80. doi:10.1126/science.1063127. PMID 11498575.
- ↑ Benevolenskaya EV (August 2007). "Histone H3K4 demethylases are essential in development and differentiation". Biochem. Cell Biol. 85 (4): 435–43. doi:10.1139/o07-057. PMID 17713579.
- ↑ 9.0 9.1 9.2 9.3 9.4 9.5 9.6 9.7 Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K (May 2007). "High-resolution profiling of histone methylations in the human genome". Cell 129 (4): 823–37. doi:10.1016/j.cell.2007.05.009. PMID 17512414.
- ↑ 10.0 10.1 10.2 Steger DJ, Lefterova MI, Ying L, Stonestrom AJ, Schupp M, Zhuo D, Vakoc AL, Kim JE, Chen J, Lazar MA, Blobel GA, Vakoc CR (April 2008). "DOT1L/KMT4 recruitment and H3K79 methylation are ubiquitously coupled with gene transcription in mammalian cells". Mol. Cell. Biol. 28 (8): 2825–39. doi:10.1128/MCB.02076-07. PMC 2293113. PMID 18285465.
- ↑ 11.0 11.1 11.2 Koch CM, Andrews RM, Flicek P, Dillon SC, Karaöz U, Clelland GK, Wilcox S, Beare DM, Fowler JC, Couttet P, James KD, Lefebvre GC, Bruce AW, Dovey OM, Ellis PD, Dhami P, Langford CF, Weng Z, Birney E, Carter NP, Vetrie D, Dunham I (June 2007). "The landscape of histone modifications across 1% of the human genome in five human cell lines". Genome Res. 17 (6): 691–707. doi:10.1101/gr.5704207. PMC 1891331. PMID 17567990.
- ↑ 12.0 12.1 Wang GG, Allis CD, Chi P. (2007). "Chromatin remodeling and cancer, Part II: ATP-dependent chromatin remodeling.". Trends Mol Med. 13 (9): 373–80. doi:10.1016/j.molmed.2007.07.004. PMID 17822959.
- ↑ Saha A, Wittmeyer J, Cairns BR (2006). "Chromatin remodelling: the industrial revolution of DNA around histones". Nat Rev Mol Cell Biol 7 (6): 437–47. doi:10.1038/nrm1945. PMID 16723979.
- ↑ Hanahan D, Weinberg RA. (2000). "The hallmarks of cancer.". Cell 100 (1): 57–70. doi:10.1016/S0092-8674(00)81683-9. PMID 10647931.
- ↑ Versteege, I; Sévenet, N; Lange, J; Rousseau-Merck, MF; Ambros, P; Handgretinger, R; Aurias, A; Delattre, O (Jul 9, 1998). "Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer.". Nature 394 (6689): 203–6. doi:10.1038/28212. PMID 9671307.
- ↑ Shain, AH; Pollack, JR (2013). "The spectrum of SWI/SNF mutations, ubiquitous in human cancers.". PLoS ONE 8 (1): e55119. doi:10.1371/journal.pone.0055119. PMC 3552954. PMID 23355908.
- ↑ Wolffe AP. (2001). "Chromatin remodeling: why it is important in cancer.". Oncogene 20 (24): 2988–90. doi:10.1038/sj.onc.1204322. PMID 11420713.
- ↑ Marks PA, Dokmanovic M (2005). "Histone deacetylase inhibitors: discovery and development as anticancer agents". Expert Opinion on Investigational Drugs 14 (12): 1497–511. doi:10.1517/13543784.14.12.1497. PMID 16307490.
- ↑ http://clincancerres.aacrjournals.org/content/8/3/662.full.pdf"Histone Deacetylase Inhibitors: A New Class of Potential Therapeutic Agents for Cancer Treatment" 2002
- ↑ Richon VM, Sandhoff TW, Rifkind RA, Marks PA (August 2000). "Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation". Proc. Natl. Acad. Sci. U.S.A. 97 (18): 10014–9. doi:10.1073/pnas.180316197. PMC 27656. PMID 10954755.
- ↑ Zhang Z, Yamashita H, Toyama T, et al. (November 2005). "Quantitation of HDAC1 mRNA expression in invasive carcinoma of the breast*". Breast Cancer Res. Treat. 94 (1): 11–6. doi:10.1007/s10549-005-6001-1. PMID 16172792.
- ↑ Dowden J, Hong W, Parry RV, Pike RA, Ward SG (April 2010). "Toward the development of potent and selective bisubstrate inhibitors of protein arginine methyltransferases". Bioorg. Med. Chem. Lett. 20 (7): 2103–5. doi:10.1016/j.bmcl.2010.02.069. PMID 20219369.
- ↑ Clapier CR, Cairns BR. (2009). "The biology of chromatin remodeling complexes.". Annu Rev Biochem. 78: 273–304. doi:10.1146/annurev.biochem.77.062706.153223. PMID 19355820.
External links
- Chromatin remodeling at the US National Library of Medicine Medical Subject Headings (MeSH)
| ||||||||||||||||||||||||||||||||||||