Charge-transfer complex
A charge-transfer complex (CT complex) or electron-donor-acceptor complex is an association of two or more molecules, or of different parts of one large molecule, in which a fraction of electronic charge is transferred between the molecular entities. The resulting electrostatic attraction provides a stabilizing force for the molecular complex. The source molecule from which the charge is transferred is called the electron donor and the receiving species is called the electron acceptor.
The nature of the attraction in a charge-transfer complex is not a stable chemical bond, and is thus much weaker than covalent forces. Many such complexes can undergo an electronic transition into an excited electronic state. The excitation energy of this transition occurs very frequently in the visible region of the electro-magnetic spectrum, which produces the characteristic intense color for these complexes. These optical absorption bands are often referred to as charge-transfer bands (CT bands). Optical spectroscopy is a powerful technique to characterize charge-transfer bands.
Charge-transfer complexes exist in many types of molecules, inorganic as well as organic, and in solids, liquids, and solutions. A well-known example is the complex formed by iodine when combined with starch, which exhibits an intense blue charge-transfer band.
In inorganic chemistry, most charge-transfer complexes involve electron transfer between metal atoms and ligands. The charge-transfer bands of transition metal complexes result from shift of charge density between molecular orbitals (MO) that are predominantly metal in character and those that are predominantly ligand in character. If the transfer occurs from the MO with ligand-like character to the metal-like one, the complex is called a ligand-to-metal charge-transfer (LMCT) complex. If the electronic charge shifts from the MO with metal-like character to the ligand-like one, the complex is called a metal-to-ligand charge-transfer (MLCT) complex. Thus, a MLCT results in oxidation of the metal center, whereas a LMCT results in the reduction of the metal center. Resonance Raman spectroscopy[2] is also a powerful technique to assign and characterize charge-transfer bands in these complexes.
Donor-acceptor association equilibrium
Charge-transfer complexes are formed by weak association of molecules or molecular subgroups, one acting as an electron donor and another as an electron acceptor. The association does not constitute a strong covalent bond and is subject to significant temperature, concentration, and host, e.g., solvent, dependencies.
The charge-transfer association occurs in a chemical equilibrium with the independent donor (D) and acceptor (A) molecules:
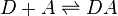
In terms of quantum mechanics, this is described as a resonance between the non-bonded state |D, A> and the dative state |D+...A->. The formation of the dative state is an electronic transition giving rise to the colorful absorption bands.
The intensity of charge-transfer bands in the absorbance spectrum is strongly dependent upon the degree (equilibrium constant) of this association reaction. Methods have been developed to determine the equilibrium constant for these complexes in solution by measuring the intensity of absorption bands as a function of the concentration of donor and acceptor components in solution. The methods were first described for the association of iodine dissolved in aromatic hydrocarbons.[3] The procedure is called the Benesi-Hildebrand method, named after the authors of the study.
Charge-transfer transition energy
The absorption wavelength of charge-transfer bands, i.e., the charge-transfer transition energy, is characteristic of the specific type of donor and acceptor entities.
The electron donating power of a donor molecule is measured by its ionization potential, which is the energy required to remove an electron from the highest occupied molecular orbital. The electron accepting power of the electron acceptor is determined by its electron affinity, which is the energy released when filling the lowest unoccupied molecular orbital.
The overall energy balance (ΔE) is the energy gained in a spontaneous charge transfer. It is determined by the difference between the acceptor's electron affinity (EA) and the donor's ionization potential (EI), adjusted by the resulting electrostatic attraction (J) between donor and acceptor:[4]

The positioning of the characteristic CT bands in the electromagnetic spectrum is directly related to this energy difference and the balance of resonance contributions of non-bonded and dative states in the resonance equilibrium.
Identification of CT bands
Charge-transfer complexes are identified by[2]
- Color: The color of CT complexes is reflective of the relative energy balance resulting from the transfer of electronic charge from donor to acceptor.
- Solvatochromism: In solution, the transition energy and therefore the complex color varies with variation in solvent permittivity, indicating variations in shifts of electron density as a result of the transition. This distinguishes it from the π* ← π transitions on the ligand.
- Intensity: CT absorptions bands are intense and often lie in the ultraviolet or visible portion of the spectrum. For inorganic complexes, the typical molar absorptivities, ε, are about 50000 L mol−1 cm−1, that are three orders of magnitude higher than typical ε of 20 L mol−1 cm−1 or lower, for d-d transitions (transition from t2g to eg). This is because the CT transitions are spin-allowed and Laporte-allowed. However, d-d transitions are only spin-allowed; they are Laporte-forbidden.
Inorganic charge-transfer complexes
Charge-transfer occurs often in inorganic ligand chemistry involving metals. Depending on the direction of charge transfer they are classified as either ligand-to-metal (LMCT) or metal-to-ligand (MLCT) charge transfer.
Ligand-to-metal charge transfer
LMCT complexes arise from transfer of electrons from MO with ligand-like character to those with metal-like character. This type of transfer is predominant if complexes have ligands with relatively high-energy lone pairs (example S or Se) or if the metal has low-lying empty orbitals. Many such complexes have metals in high oxidation states (even d0). These conditions imply that the acceptor level is available and low in energy.
Consider a d6 octahedral complex, such as IrBr63-), whose t2g levels are filled. As a consequence, an intense absorption is observed around 250 nm corresponding to a transition from ligand σ MO to the empty eg MO. However, in IrBr62- that is a d5 complex two absorptions, one near 600 nm and another near 270 nm, are observed. This is because two transitions are possible, one to t2g (that can now accommodate one more electron) and another to eg. The 600 nm band corresponds to transition to the t2g MO and the 270 nm band to the eg MO.
Charge transfer bands may also arise from transfer of electrons from nonbonding orbitals of the ligand to the eg MO.
Trend of LMCT energies
- Oxidation Number
- +7 MnO4- < TcO4- < ReO4-
- +6 CrO42- < MoO42- < WO42-
- +5 VO43- < NbO43- < TaO43-
The energies of transitions correlate with the order of the electrochemical series. The metal ions that are most easily reduced correspond to the lowest energy transitions. The above trend is consistent with transfer of electrons from the ligand to the metal, thus resulting in a reduction of metal ions by the ligand.
Examples include:
- MnO4- : The permanganate ion having tetrahedral geometry is intensely purple due to strong absorption involving charge transfer from MO derived primarily from filled oxygen p orbitals to empty MO derived from manganese(VII).
- CdS: The color of artist’s pigment cadmium-yellow is due to transition from Cd2+ (5s) ← S2-(π).
- HgS: it is red due to Hg2+ (6s) ← S2-(π) transition.
- Fe Oxides: they are red and yellow due to transition from Fe (3d) ← O2-(π).
Metal-to-ligand charge transfer
Metal-to-ligand charge-transfer (MLCT) complexes arise from transfer of electrons from MO with metal-like character to those with ligand-like character.[2][5] This is most commonly observed in complexes with ligands having low-lying π* orbitals, especially aromatic ligands. The transition will occur at low energy if the metal ion has a low oxidation number, for its d orbitals will relatively be high in energy.
Examples of such ligands taking part in MLCT include 2,2'-bipyridine (bipy), 1,10-phenanthroline (phen), CO, CN- and SCN-. Examples of these complexes include:
- Tris(2,2’-bipyridyl)ruthenium(II) : This orange-color complex is being studied,[6] as the excited state resulting from this charge transfer has a lifetime of microseconds and the complex is a versatile photochemical redox reagent.
- W(CO)4(phen)
- Fe(CO)3(bipy)
Photoreactivity of MLCT excited states
The photoreactivity of MLCT complexes result from the nature of the oxidized metal and the reduced ligand. Though the states of traditional MLCT complexes like Ru(bipy)32+ and Re(bipy)(CO)3Cl were intrinsically not reactive, several MLCT complexes that are characterized by reactive MLCT states have been synthesized.
Vogler and Kunkely[7] considered the MLCT complex to be an isomer of the ground state, which contains an oxidized metal and reduced ligand. Thus, various reactions like electrophilic attack and radical reactions on the reduced ligand, oxidative addition at the metal center due to the reduced ligand, and outer sphere charge-transfer reactions can be attributed to states arising from MLCT transitions. MLCT states’ reactivity often depends on the oxidation of the metal. Subsequent processes include associative ligand substitution, exciplex formation, and cleavage of metal---metal bonds.
Color of charge-transfer complexes
- The spin rule: Δ S = 0
On promotion, the electron should not experience a change in spin. Electronic transitions that experience a change in spin are said to be spin-forbidden.
- Laporte's rule: Δ l = ± 1
d-d transitions for complexes that have a center of symmetry are forbidden - symmetry-forbidden or Laporte-forbidden.[8]
Charge-transfer complexes do not experience d-d transitions. Thus, these rules do not apply and, in general, the absorptions are very intense.
For example, the classic example of a charge-transfer complex is that between iodine and starch to form an intense purple color. This has widespread use as a rough screen for counterfeit currency. Unlike most paper, the paper used in US currency is not sized with starch. Thus, formation of this purple color on application of an iodine solution indicates a counterfeit.
Other examples
Hexaphenylbenzenes like H (Fig. 2) lend themselves extremely well to forming charge-transfer complexes. Cyclic voltammetry for H displays 4 well-separated maxima corresponding to H+ right up to H4+ with the first ionization at E1/2 of only 0.51 eV. oxidation of these arenes by for instance dodecamethylcarboranyl (B) to the blue crystal solid H+B- complex is therefore easy.[9]

The phenyl groups are all positioned in an angle of around 45° with respect to the central aromatic ring and the positive charge in the radical cation is therefore through-space-delocalised through the 6 benzene rings in the shape of a toroid. The complex has 5 absorption bands in the near-infrared region, which can be assigned to specific electronic transitions with the aid of deconvolution and the Mulliken-Hush theory.
Electrical conductivity
In 1954, researchers at Bell Laboratories and elsewhere reported charge-transfer complexes with resistivities as low as 8 ohms·cm in combinations of perylene with iodine or bromine.[10][11] In 1962, the well-known acceptor tetracyanoquinodimethane (TCNQ) was reported. Tetrathiafulvalene (TTF) was synthesized in 1970 and found to be a strong electron donor. In 1973, it was discovered that a combination of these components form a strong charge-transfer complex, henceforth referred to as TTF-TCNQ.[12] The complex is formed in solution and may be crystallized into a well-formed crystalline solid. The solid shows almost metallic electrical conductance and was the first discovered purely organic conductor. In a TTF-TCNQ crystal, TTF and TCNQ molecules are arranged independently in separate parallel-aligned stacks, and an electron transfer occurs from donor (TTF) to acceptor (TCNQ) stacks. Hence, electrons and electron holes are separated and concentrated in the stacks and can traverse in a one-dimensional direction along the TCNQ and TTF columns, respectively, when an electric potential is applied to the ends of a crystal in the stack direction.
The first organic molecule that forms a superconductor was discovered in 1980. Tetramethyl-tetraselenafulvalene-phosphorus hexafluoride (TMTSF2PF6), a semi-conductor at ambient conditions, shows superconductivity at low temperature (critical temperature) and high pressure: 0.9 K and 12 kbar. Since 1980, many organic superconductors have been synthesized, and the critical temperature has been raised to over 100 K as of 2001.[citation needed] Unfortunately, critical current densities in these complexes are very small.
See also
References
- ↑ Housecroft, C. E.; Sharpe, A. G. (2008). Inorganic Chemistry (3rd ed.). Prentice Hall. p. 541. ISBN 978-0131755536.
- ↑ 2.0 2.1 2.2 Atkins, P. J.; Shriver, D. F. (1999). Inorganic chemistry (3rd ed.). New York: W.H. Freeman and CO. ISBN 0-7167-3624-1.
- ↑ H. Benesi, J. Hildebrand, A Spectrophotometric Investigation of the Interaction of Iodine with Aromatic Hydrocarbons, J. Am. Chem. Soc. 71(8), 2703-07 (1949).
- ↑ Mulliken, R. S.; Person, W. B. (1969). Molecular Complexes. New York and London: Wiley-Interscience. doi:10.1016/0022-2860(71)87071-0. ISBN 0-471-62370-9.
- ↑ Tarr, Donald A.; Miessler, Gary L. (1991). Inorganic chemistry (2nd ed.). Englewood Cliffs, N.J: Prentice Hall. ISBN 0-13-465659-8.
- ↑ Kalyanasundaram, K. (1992). Photochemistry of polypyridine and porphyrin complexes. Boston: Academic Press. ISBN 0-12-394992-0.
- ↑ Vogler, A.; Kunkely, H. (2000). "Photochemistry induced by metal-to-ligand charge transfer excitation". Coord. Chem. Rev. 208: 321. doi:10.1016/S0010-8545(99)00246-5.
- ↑ Robert J. Lancashire. "Selection rules for Electronic Spectroscopy". University of the West Indies, Mona. Retrieved 2008-08-30.
- ↑ Duoli Sun, Sergiy V. Rosokha, Jay K. Kochi (2005). "Through-Space (Cofacial) -Delocalization among Multiple Aromatic Centers: Toroidal Conjugation in Hexaphenylbenzene-like Radical Cations". Angew. Chem. Int. Ed. 44 (32): 5133–5136. doi:10.1002/anie.200501005.
- ↑ Y. Okamoto and W. Brenner Organic Semiconductors, Rheinhold (1964)
- ↑ H. Akamatsu, H. Inokuchi, and Y.Matsunaga (1954). "Electrical Conductivity of the Perylene–Bromine Complex". Nature 173 (4395): 168. Bibcode:1954Natur.173..168A. doi:10.1038/173168a0.
- ↑ P. W. Anderson, P. A. Lee, M. Saitoh (1973). "Remarks on giant conductivity in TTF-TCNQ". Solid State Communications 13: 595–598. Bibcode:1973SSCom..13..595A. doi:10.1016/S0038-1098(73)80020-1.