Carbon–hydrogen bond activation
Carbon–hydrogen bond activation or C–H activation is a reaction that cleaves a carbon–hydrogen bond.[1][2][3][4][5][6][7][8][9][10][11] The term is often restricted to reactions that involve organometallic complexes and transformations that proceed by coordination of a hydrocarbon to the inner-sphere of a metal, either via an intermediate “alkane or arene complex” or as a transition state leading to a "M−C" intermediate.[12][13][14] Important to this definition is the requirement that during the C–H cleavage event, the hydrocarbyl species remains associated in the inner-sphere and under the influence of “M”.
Theoretical studies as well as experimental investigations indicate that C–H bonds, which are traditionally considered unreactive, can be cleaved by coordination. Much research effort has been devoted to the design and synthesis of new reagents and catalysts that can affect C–H activation. A significant driver for this type of research is the prospect that C–H activation could enable the conversion of cheap and abundant alkanes into valuable functionalized organic compounds and the efficient structural editing of already complex molecules.[15]
Historic overview
The first C–H activation reaction is often attributed to Otto Dimroth, who in 1902, reported that benzene reacted with mercury(II) acetate (See: organomercury), but some scholars do not view this reaction as being true C–H activation. As observed by Goldman & Goldberg[12] C–H activation resembles aspects of H−H activation: both can be achieved by electrophilic or oxidative addition.
From a modern organometallic perspective, the first true C–H activation reaction was reported by Joseph Chatt in 1965[16] with insertion of a ruthenium atom ligated to dmpe in the C–H bond of naphthalene. In 1969, A.E. Shilov reported that potassium tetrachloroplatinate induced isotope scrambling between methane and heavy water. The pathway was proposed to involve binding of methane to Pt(II). In 1972, the Shilov group was able to produce methanol and methyl chloride in a similar reaction involving a stoichiometric amount of potassium tetrachloroplatinate, catalytic potassium hexachloroplatinate, methane and water. Due to the fact that Shilov worked and published in the Soviet Union during the Cold War era, his work was largely ignored by Western scientists. This so-called Shilov system is today one of the few true catalytic systems for alkane functionalizations.[12]
On the other side of the spectrum, oxidative addition, M. L. H. Green in 1970 reported on the photochemical insertion of tungsten (as a Cp2WH2 complex) in a benzene C–H bond[17] and George M. Whitesides in 1979 was the first to carry out an intramolecular aliphatic C–H activation[18]
The next breakthrough was reported independently by two research groups in 1982. R. G. Bergman reported the first transition metal-mediated intermolecular C–H activation of unactivated and completely saturated hydrocarbons by oxidative addition. Using a photochemical approach, photolysis of Cp*Ir(PMe3)H2, where Cp* is a pentamethylcyclopentadienyl ligand, led to the coordinatively unsaturated species Cp*Ir(PMe3) which reacted via oxidative addition with cyclohexane and neopentane to form the corresponding hydridoalkyl complexes, Cp*Ir(PMe3)HR, where R = cyclohexyl and neopentyl, respectively.[19] W.A.G. Graham found that the same hydrocarbons react with Cp*Ir(CO)2 upon irradiation to afford the related alkylhydrido complexes Cp*Ir(CO)HR, where R = cyclohexyl and neopentyl, respectively.[20] In the latter example, the reaction is presumed to proceed via the oxidative addition of alkane to a 16-electron iridium(I) intermediate, Cp*Ir(CO), formed by irradiation of Cp*Ir(CO)2.
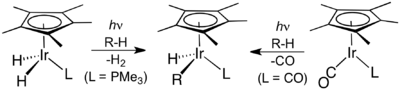 C–H activation by Bergman et al. (left) and Graham et al.
C–H activation by Bergman et al. (left) and Graham et al.
J.F. Hartwig reported a highly regioselective arene and alkane borylation catalyzed by a rhodium complex in 1999 and 2000. In the case of alkanes, exclusive terminal functionalization was observed.[21]
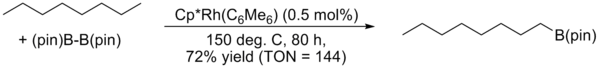
Scope
The selective activation and functionalization of alkane C–H bonds was reported using a tungsten complex outfitted with pentamethylcyclopentadienyl, nitrosyl, allyl and neopentyl ligands, Cp*W(NO)(η3-allyl)(CH2CMe3).[22]
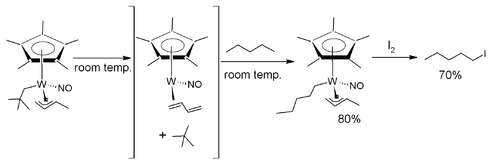
In one example involving this system, the alkane pentane is selectively converted to the halocarbon 1-iodopentane. This transformation was achieved via the thermoloysis of Cp*W(NO)(η3-allyl)(CH2CMe3) in pentane at room temperature, resulting in elimination of neopentane by a pseudo-first-order process, generating an undetectable electronically and sterically unsaturated 16-electron intermediate that is coordinated by an η2-butadiene ligand. Subsequent intermolecular activation of a pentane solvent molecule then yields an 18-electron complex possessing an n-pentyl ligand. In a separate step, reaction with iodine at −60 °C liberates 1-iodopentane from the complex.
Arene C–H bonds can also be activated by metal complexes despite being fairly unreactive. One manifestation is found in the Murai olefin coupling.[23] In one reaction a ruthenium complex reacts with N,N-dimethylbenzylamine in a cyclometalation also involving C–H activation:[24]

An alkene C–H bond activation with a rhodium catalyst is demonstrated in the synthesis of this strained bicyclic enamine:[25]

It has also been found by Prof. Roy A. Periana that reactions of late transition metal salts and complexes such of Pt, Pd, Au, and Hg react with methane (CH4), the major component of natural gas, in H2SO4 to yield methyl bisulfate in very low to high yields at very high selectivity.[26][27]
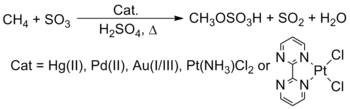
Reaction conditions
Most C–H bond activations proceed under rather harsh reaction conditions (high T, strongly acidic or basic conditions, strong oxidant), significantly lowering their attractiveness. However, more and more quite mild reactions have been developed, significantly expanding the scope of these exciting transformations.[28] Organocatalysis has recently been used as an important approach which has the advantage of being metal-free and cost-effective.[29]
Recently, Hashiguchi et al. discovered that M-hydroxides and aquo complexes can be utilized to carry out C–H activation on water soluble arenes in strongly basic conditions.[30] Specifically, they demonstrated a Ru precatalyst with an NNN-pincer ligand, 2,6-diimidizoylpyridine, (IPI), when dissolved in aqueous base generated the associated mixed hydroxide aquo complex, Ru(IPI(OH)n(H2O)m. This complex was then shown to catalyze H/D exchange between the arenes and aqueous base with increasing [KOH] upon initial reduction of Ru(III) to the Ru(II) complex. This was the first demonstrated report of base accelerated CH activation.

Metal centers that contain a large number of electrons and are stable at high oxidation states, such as later transition metals, promote oxidative addition and are therefore ideal candidates to act as catalysts for C–H activation. Furthermore d8 metals that can undergo a square planar arrangement, are potential candidates catalysts as they have two vacant coordination sites and thus steric hindrance is limited.
See also
References
- ↑ “Alkane C–H activation and functionalization with homogeneous transition metal catalysts: a century of progress – a new millennium in prospect”, R. H. Crabtree, J. Chem. Soc., Dalton Trans. 2001, 17, 2437–2450. doi:10.1039/B103147N
- ↑ "Designing Catalysts for Functionalization of Unactivated C–H Bonds Based on the CH Activation Reaction" B. G. Hashiguchi, S. M. Bischof, M. M. Konnick, R. A. Periana. Acc. Chem. Res. 2012, 45, 885–898. doi:10.1021/ar200250r
- ↑ “Organometallic alkane CH activation”, R. H. Crabtree, J. Organomet. Chem. 2004, 689, 4083–4091. doi:10.1016/j.jorganchem.2004.07.034
- ↑ “Mechanistic Aspects of C−H Activation by Pt Complexes”, M. Lersch, M.Tilset, Chem. Rev. 2005, 105, 2471–2526. doi:10.1021/cr030710y
- ↑ “Recent Advances in the Platinum-mediated CH Bond Functionalization”, A. N. Vedernikov, Curr. Org. Chem. 2007, 11, 1401–1416.
- ↑ “Catalytic C–H functionalization by metalcarbenoid and nitrenoid insertion”, H. M. L. Davies, J. R. Manning, Nature, 2008, 451, 417–424, doi:10.1038/nature06485
- ↑ "Mechanisms of C–H bond activation: rich synergy between computation and experiment”, Y. Boutadla, D. L. Davies, S. A. Macgregor, A. I. Poblador-Bahamonde, Dalton Trans. 2009, 5820–5831. doi:10.1039/B904967C
- ↑ “C–H Bond Activation in Transition Metal Species from a Computational Perspective”, D. Balcells, E. Clot, O. Eisenstein, Chem. Rev. 2010, 110, 749–823. doi:10.1021/cr900315k
- ↑ “Palladium-Catalyzed Ligand-Directed C–H Functionalization Reactions”, T. W. Lyons, M. S. Sanford, Chem. Rev. 2010, 110, 1147–1169. doi:10.1021/cr900184e
- ↑ “Beyond Directing Groups: Transition Metal-Catalyzed C H Activation of Simple Arenes”, N. Kuhl, M. N. Hopkinson, J. Wencel-Delord, F. Glorius, Angew. Chem. Int. Ed. 2012, 51, 10236–10254. doi:10.1002/anie.201203269
- ↑ “Selectivity enhancement in functionalization of C–H bonds: A review”, G. B. Shul’pin, Org. Biomol. Chem. 2010, 8, 4217–4228. doi:10.1039/c004223d
- ↑ 12.0 12.1 12.2 Organometallic C–H Bond Activation: An Introduction Alan S. Goldman and Karen I. Goldberg ACS Symposium Series 885, Activation and Functionalization of C–H Bonds, 2004, 1–43
- ↑ Arndtsen, B. A.; Bergman, R. G.; Mobley, T. A.; Peterson, T. H. “Selective Intermolecular Carbon–Hydrogen Bond Activation by Synthetic Metal Complexes in Homogeneous Solution.” Accounts of Chemical Research, 1995: 28 (3) 154–162.
- ↑ Periana, R. A.; Bhalla, G.; Tenn, W. J., III, Young, K. J. H.; Liu, X. Y.; Mironov, O.; Jones, C.; Ziatdinov, V. R. “Perspectives on some challenges and approaches for developing the next generation of selective, low temperature, oxidation catalysts for alkane hydroxylation based on the C–H activation reaction.” Journal of Molecular Catalysis A: Chemical, 2004: 220 (1) 7–25. doi:10.1016/j.molcata.2004.05.036
- ↑ “C–H bond activation enables the rapid construction and late-stage diversification of functional molecules”, J. Wencel-Delord, F. Glorius, Nature Chem. 2013, 5, 369–375. doi:10.1038/nchem.1607
- ↑ The tautomerism of arene and ditertiary phosphine complexes of ruthenium(0), and the preparation of new types of hydrido-complexes of ruthenium(II) J. Chatt and J. M. Davidson, J. Chem. Soc. 1965, 843 doi:10.1039/JR9650000843
- ↑ Formation of a tangsten phenyl hydride derivatives from benzene M. L. Green, P. J. Knowles, J. Chem. Soc. D, 1970, (24),1677–1677 doi:10.1039/C29700001677
- ↑ Thermal generation of bis(triethylphosphine)-3,3-dimethylplatinacyclobutane from dineopentylbis(triethylphosphine)platinum(II) Paul Foley, George M. Whitesides J. Am. Chem. Soc. 1979; 101(10); 2732–2733. doi:10.1021/ja00504a041
- ↑ Carbon–hydrogen activation in saturated hydrocarbons: direct observation of M + R−H → M(R)(H) Andrew H. Janowicz, Robert G. Bergman J. Am. Chem. Soc.; 1982; 104(1); 352–354.doi:10.1021/ja00365a091
- ↑ Oxidative addition of the carbon–hydrogen bonds of neopentane and cyclohexane to a photochemically generated iridium(I) complex James K. Hoyano, William A. G. Graham J. Am. Chem. Soc. 1982; 104(13); 3723–3725. doi:10.1021/ja00377a032
- ↑ Thermal, Catalytic, Regiospecific Functionalization of Alkanes Huiyuan Chen, Sabine Schlecht, Thomas C. Semple, John F. Hartwig Science 2000; 287(5460); 1995–1997. doi:10.1126/science.287.5460.1995
- ↑ Baillie, Rhett A.; Legzdins, Peter (2013). "Distinctive Activation and Functionalization of Hydrocarbon C–H Bonds Initiated by Cp*W(NO)(η3-allyl)(CH2CMe3) Complexes". Acc. Chem. Res: ASAP. doi:10.1021/ar400108p.
- ↑ Murai, Shinji; Kakiuchi, Fumitoshi; Sekine, Shinya; Tanaka, Yasuo; Kamatani, Asayuki; Sonoda, Motohiro; Chatani, Naoto (1993). "Efficient catalytic addition of aromatic carbon–hydrogen bonds to olefins". Nature 366 (6455): 529–531. Bibcode:1993Natur.366..529M. doi:10.1038/366529a0.
- ↑ Formation of a Ruthenium–Arene Complex, Cyclometallation with a Substituted Benzylamine, and Insertion of an Alkyne Chetcuti, Michael J.; Ritleng, Vincent. J. Chem. Educ. 2007, 84, 1014. Abstract
- ↑ The Stereoselective Formation of Bicyclic Enamines with Bridgehead Unsaturation via Tandem C–H Bond Activation/Alkenylation/ Electrocyclization Sirilata Yotphan, Robert G. Bergman, and Jonathan A. Ellman J. Am. Chem. Soc. 2008, 130, 2452–2453 doi:10.1021/ja710981b
- ↑ Periana, R.A.; Taube, D.J.; Evitt, E.R.; Loffler, D.G.; Wentrcek, P.R.; Voss, G.; Masuda, T. (1993). "A Mercury-Catalyzed, High-Yield System for the Oxidation of Methane to Methanol". Science 259 (5093): 340–343. doi:10.1126/science.280.5363.493f. PMID 17832346.
- ↑ Periana, R. A.; Taube, D. J.; Gamble, S.; Taube, H.; Satoh, T.; Fujii, H. (1998). "Platinum Catalysts for the High-Yield Oxidation of Methane to a Methanol Derivative". Science 280 (5363): 560–564. Bibcode:1998Sci...280..560P. doi:10.1126/science.280.5363.560. PMID 9554841.
- ↑ Towards Mild Metal-Catalyzed C–H Bond Activation J. Wencel-Delord, T. Dröge, F. Liu, F. Glorius Chem. Soc. Rev. 2011, 40, 4740–4761 doi:10.1039/C1CS15083A
- ↑ Pan, S. C. “Organocatalytic C–H activation reactions” Beilstein J. Org. Chem. 2012, 8, 1374–1384. doi:10.3762/bjoc.8.159 (Open Access)
- ↑ Acceleration of Nucleophilic CH Activation by Strongly Basic Solvents B. G. Hashiguchi, K. J. H. Young, Muhammed Yousufuddin, W. A. Goddard, III, R. A. Periana J. Am. Chem. Soc. 2010, 132, 12542-12545 doi:10.1021/ja102518m