Position-specific scoring matrix
A position weight matrix (PWM), also called position-specific weight matrix (PSWM) or position-specific scoring matrix (PSSM), is a commonly used representation of motifs (patterns) in biological sequences.[1]
A PWM is a matrix of score values that gives a weighted match to any given substring of fixed length. It has one row for each symbol of the alphabet, and one column for each position in the pattern. The score assigned by a PWM to a substring 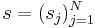 is defined as
is defined as 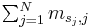 , where
, where 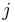 represents position in the substring,
represents position in the substring, 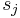 is the symbol at position in the substring, and
is the symbol at position in the substring, and 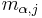 is the score in row
is the score in row 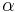 , column of the matrix. In other words, a PWM score is the sum of position-specific scores for each symbol in the substring.
, column of the matrix. In other words, a PWM score is the sum of position-specific scores for each symbol in the substring.
Contents |
Basic PWM with log-likelihoods
A PWM assumes independence between positions in the pattern, as it calculates scores at each position independently from the symbols at other positions. The score of a substring aligned with a PWM can be interpreted as the log-likelihood of the substring under a product multinomial distribution. Since each column defines log-likelihoods for each of the different symbols, where the sum of likelihoods in a column equals one, the PWM corresponds to a Multinomial distribution. A PWM's score is the sum of log-likelihoods, which corresponds to the product of likelihoods, meaning that the score of a PWM is then a product-multinomial distribution. The PWM scores can also be interpreted in a physical framework as the sum of binding energies for all nucleotides (symbols of the substring) aligned with the PWM.
Incorporating background distribution
Instead of using log-likelihood values in the PWM, as described in the previous paragraph, several methods uses log-odds scores in the PWMs. An element in a PWM is then calculated as 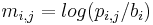 , where
, where 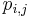 is the probability of observing symbol i at position j of the motif, and
is the probability of observing symbol i at position j of the motif, and 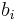 is the probability of observing the symbol i in a background model. The PWM score then corresponds to the log-odds of the substring being generated by the motif versus being generated by the background, in a generative model of the sequence.
is the probability of observing the symbol i in a background model. The PWM score then corresponds to the log-odds of the substring being generated by the motif versus being generated by the background, in a generative model of the sequence.
Information content of a PWM
The information content (IC) of a PWM is sometimes of interest, as it says something about how different a given PWM is from a uniform distribution.
The self-information of observing a particular symbol at a particular position of the motif is:
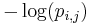
The expected (average) self-information of a particular element in the PWM is then:
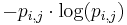
Finally, the IC of the PWM is then the sum of the expected self-information of every element:
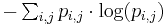
Often, it is more useful to calculate the information content with the background letter frequencies of the sequences you are studying rather than assuming equal probabilities of each letter (e.g., the GC-content of DNA of thermophilic bacteria range from 65.3 to 70.8,[2] thus a motif of ATAT would contain much more information than a motif of CCGG). The equation for information content thus becomes
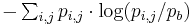
where 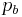 is the background frequency for that letter. This corresponds to the Kullback-Leibler divergence or relative entropy. However, it has been shown that when using PSSM to search genomic sequences (see below) this uniform correction can lead to overestimation of the importance of the different bases in a motif, due to the uneven distribution of n-mers in real genomes, leading to a significantly larger number of false positives.[3]
is the background frequency for that letter. This corresponds to the Kullback-Leibler divergence or relative entropy. However, it has been shown that when using PSSM to search genomic sequences (see below) this uniform correction can lead to overestimation of the importance of the different bases in a motif, due to the uneven distribution of n-mers in real genomes, leading to a significantly larger number of false positives.[3]
Using PWMs
There are various algorithms to scan for hits of PWMs in sequences. One example is the MATCH algorithm[4] which has been implemented in the ModuleMaster.[5] More sophisticated algorithms for fast database searching with nucleotide as well as amino acid PWMs/PSSMs are implemented in the possumsearch software and are described by Beckstette, et al. (2006).[6]
References
- ^ Ben-Gal I, Shani A, Gohr A, Grau J, Arviv S, Shmilovici A, Posch S, Grosse I (2005). "Identification of Transcription Factor Binding Sites with Variable-order Bayesian Networks". Bioinformatics 21 (11): 2657–2666. doi:10.1093/bioinformatics/bti410. PMID 15797905. http://bioinformatics.oxfordjournals.org/cgi/reprint/bti410?ijkey=KkxNhRdTSfvtvXY&keytype=ref.
- ^ Aleksandrushkina NI, Egorova LA (1978). "Nucleotide makeup of the DNA of thermophilic bacteria of the genus Thermus". Mikrobiologiia 47 (2): 250–2. PMID 661633.
- ^ Erill I, O'Neill MC (2009). "A reexamination of information theory-based methods for DNA-binding site identification". BMC Bioinformatics 10: 57. doi:10.1186/1471-2105-10-57. PMC 2680408. PMID 19210776. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2680408.
- ^ Kel AE, et. al. (2003). "MATCHTM: a tool for searching transcription factor binding sites in DNA sequences". Nucleic Acids Research 31 (13): 3576–3579. doi:10.1093/nar/gkg585. PMC 169193. PMID 12824369. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=169193.
- ^ Wrzodek, Clemens; Schröder, Adrian; Dräger, Andreas; Wanke, Dierk; Berendzen, Kenneth W.; Kronfeld, Marcel; Harter, Klaus; Zell, Andreas (9 October 2009). "ModuleMaster: A new tool to decipher transcriptional regulatory networks". Biosystems (Ireland: Elsevier) 99 (1): 79–81. doi:10.1016/j.biosystems.2009.09.005. ISSN 0303-2647. PMID 19819296.
- ^ Beckstette, M. et al. (2006). "Fast index based algorithms and software for matching position specific scoring matrices". BMC Bioinformatics 7: 389. doi:10.1186/1471-2105-7-389. PMC 1635428. PMID 16930469. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1635428.