Chemical similarity
Chemical similarity (or molecular similarity) refers to the similarity of chemical elements, molecules or chemical compounds with respect to either structural or functional qualities, i.e. the effect that the chemical compound has on reaction partners in anorganic or biological settings. Biological effects and thus also similarity of effects are usually quantified using the biological activity of a compound. In general terms, function can be related to the chemical activity of compounds (among others).
The notion of chemical similarity (or molecular similarity) is one of the most important concepts in chemoinformatics.[1][2] It plays an important role in modern approaches to predicting the properties of chemical compounds, designing chemicals with a predefined set of properties and, especially, in conducting drug design studies by screening large databases containing structures of available (or potentially available) chemicals. These studies are based on the similar property principle of Johnson and Maggiora, which states: similar compounds have similar properties.[1]
Contents |
Similarity Measures
Chemical similarity is often described as an inverse of a measure of distance in descriptor space. Distance measures can be classified into Euclidean measures and non-Euclidean measures depending on whether the triangle inequality holds.
Similarity Search and Virtual Screening
The similarity-based [3] virtual screening (a kind of ligand-based virtual screening) assumes that all compounds in a database that are similar to a query compound have similar biological activity. Although this hypothesis is not always valid,[4] quite often the set of retrieved compounds is considerably enriched with actives.[5] To achieve high efficacy of similarity-based screening of databases containing millions of compounds, molecular structures are usually represented by molecular screens (structural keys) or by fixed-size or variable-size molecular fingerprints. Molecular screens and fingerprints can contain both 2D- and 3D-information. However, the 2D-fingerprints, which are a kind of binary fragment descriptors, dominate in this area. Fragment-based structural keys, like MDL keys,[6] are sufficiently good for handling small and medium-sized chemical databases, whereas processing of large databases is performed with fingerprints having much higher information density. Fragment-based Daylight,[7] BCI,[8] and UNITY 2D (Tripos[9]) fingerprints are the best known examples. The most popular similarity measure for comparing chemical structures represented by means of fingerprints is the Tanimoto (or Jaccard) coefficient T. Two structures are usually considered similar if 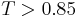 (for Daylight fingerprints).
(for Daylight fingerprints).
References
- ^ a b A. M. Johnson, G. M. Maggiora (1990). Concepts and Applications of Molecular Similarity. New York: John Willey & Sons. ISBN 0471621757.
- ^ N. Nikolova, J. Jaworska (2003). "Approaches to Measure Chemical Similarity - a Review". QSAR & Combinatorial Science 22 (9-10): 1006–1026. doi:10.1002/qsar.200330831.
- ^ S. A. Rahman, M. Bashton, G. L. Holliday, R. Schrader and J. M. Thornton, Small Molecule Subgraph Detector (SMSD) toolkit, Journal of Cheminformatics 2009, 1:12. DOI:10.1186/1758-2946-1-12
- ^ H. Kubinyi (1998). "Similarity and Dissimilarity: A Medicinal Chemist’s View". Persp. Drug Discov. Design 9-11: 225–252. doi:10.1023/A:1027221424359.
- ^ Y. C. Martin, J. L. Kofron, L. M. Traphagen (2002). "Do structurally similar molecules have similar biological activity?". J. Med. Chem. 45 (19): 4350–4358. doi:10.1021/jm020155c. PMID 12213076.
- ^ J. L. Durant, B. A. Leland, D. R. Henry, J. G. Nourse (2002). "Reoptimization of MDL Keys for Use in Drug Discovery". J. Chem. Inf. Comput. Sci. 42 (6): 1273–1280. PMID 12444722.
- ^ "Daylight Chemical Information Systems Inc.". http://www.daylight.com.
- ^ "Barnard Chemical Information Ltd.". http://www.bci.gb.com/.
- ^ "Tripos Inc.". http://www.tripos.com.
External links
- Small Molecule Subgraph Detector (SMSD) - is a Java based software library for calculating Maximum Common Subgraph (MCS) between small molecules. This will help us to find similarity/distance between two molecules. MCS is also used for screening drug like compounds by hitting molecules, which share common subgraph (substructure).
- Chemical Similarity (QSAR World)
- Similarity Principle
- Fingerprint-based Similarity used in QSAR Modeling
- Brutus is a similarity analysis tool based on molecular interaction fields.