Telomerase
| RNA-directed DNA polymerase | |||||||
|---|---|---|---|---|---|---|---|
 |
|||||||
| A conceptual diagram showing the protein component of telomerase (TERT) in grey and the RNA component (TR) in yellow | |||||||
| Identifiers | |||||||
| EC number | 2.7.7.49 | ||||||
| CAS number | 9068-38-6 | ||||||
| Databases | |||||||
| IntEnz | IntEnz view | ||||||
| BRENDA | BRENDA entry | ||||||
| ExPASy | NiceZyme view | ||||||
| KEGG | KEGG entry | ||||||
| MetaCyc | metabolic pathway | ||||||
| PRIAM | profile | ||||||
| PDB | structures | ||||||
| Gene Ontology | AmiGO / EGO | ||||||
|
|||||||
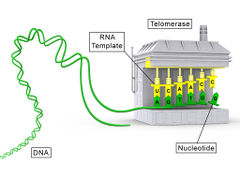
Telomerase is an enzyme that adds DNA sequence repeats ("TTAGGG" in all vertebrates) to the 3' end of DNA strands in the telomere regions, which are found at the ends of eukaryotic chromosomes. This region of repeated nucleotide repeats called telomeres contain condensed DNA material and prevents constant loss of important DNA from chromosome ends. As a result, every time the chromosome is copied only a couple telomeres are lost, which causes no damage to the organism. Telomerase is a reverse transcriptase that carries its own RNA molecule, which is used as a template when it elongates telomeres, which are shortened after each replication cycle. The existence of a compensatory shortening of telomere (telomerase) mechanism was first predicted by Soviet biologist Alexey Olovnikov in 1973,[1] who also suggested the telomere hypothesis of aging and the telomere's connections to cancer. Telomerase was discovered by Carol W. Greider and Elizabeth Blackburn in 1984 in the ciliate Tetrahymena.[2] Together with Jack W. Szostak, Greider and Blackburn were awarded the 2009 Nobel Prize in Physiology or Medicine for their discovery.[3]
Contents |
Structure
The protein composition of human telomerase was identified in 2007 by Scott Cohen and his team at the Children's Medical Research Institute in Australia. It consists of two molecules each of human telomerase reverse transcriptase (TERT), telomerase RNA (TR or TERC), and dyskerin (DKC1).[4] The genes of telomerase subunits, which are TERT,[5] TERC,[6] DKC1,[7] and TEP1[8] etc, are located on the different chromosomes in human genome. Human TERT gene (hTERT) is translated into a protein of 1132 amino acids.[9] TERT proteins from many eukaryotes have been sequenced.[10] TERT polypeptide folds with TERC, a non-coding RNA (451 nucleotides long in human). TERT has a 'mitten' structure that allows it to wrap around the chromosome to add single-stranded telomere repeats.
TERT is a reverse transcriptase, which is a class of enzyme that creates single-stranded DNA using single-stranded RNA as a template. Enzymes of this class (not TERT specifically, but the ones isolated from viruses) are utilized by scientists in the molecular biological process of reverse transcriptase PCR (RT-PCR), which allows the creation of several DNA copies of a target sequence using RNA as a template. As stated above, TERT carries its own template around, TERC.
The high-resolution protein structure of the Tribolium castaneum catalytic subunit of telomerase TERT was decoded in 2008 by Emmanuel Skordalakes and his team at The Wistar Institute in Philadelphia.[11] The structure revealed that the protein consists of four conserved domains (RNA-Binding Domain (TRBD), fingers, palm and thumb), organized into a ring configuration that shares common features with retroviral reverse transcriptases, viral RNA polymerases and bacteriophage B-family DNA polymerases.
Function
By using TERC, TERT can add a six-nucleotide repeating sequence, 5'-TTAGGG (in all vertebrates, the sequence differs in other organisms) to the 3' strand of chromosomes. These TTAGGG repeats (with their various protein binding partners) are called telomeres. The template region of TERC is 3'-CAAUCCCAAUC-5'.[12] This way, telomerase can bind the first few nucleotides of the template to the last telomere sequence on the chromosome, add a new telomere repeat (5'-GGTTAG-3') sequence, let go, realign the new 3'-end of telomere to the template, and repeat the process. (For an explanation on why this elongation is necessary see Telomere shortening.)
Clinical implications
Aging
The enzyme telomerase allows for replacement of short bits of DNA known as telomeres, which are otherwise shortened when a cell divides via mitosis.
In normal circumstances, without the presence of telomerase, if a cell divides recursively, at some point all the progeny will reach their Hayflick limit.[13] With the presence of telomerase, each dividing cell can replace the lost bit of DNA, and any single cell can then divide unbounded. While this unbounded growth property has excited many researchers, caution is warranted in exploiting this property, as exactly this same unbounded growth is a crucial step in enabling cancerous growth.
Embryonic stem cells express telomerase, which allows them to divide repeatedly and form the individual. In adults, telomerase is highly expressed in cells that need to divide regularly (e.g., in the immune system), whereas most somatic cells express it only at very low levels in a cell-cycle-dependent manner.
A variety of premature aging syndromes are associated with short telomeres.[14] These include Werner syndrome, Ataxia telangiectasia, Ataxia-telangiectasia like disorder, Bloom syndrome, Fanconi anemia and Nijmegen breakage syndrome. The genes that have been mutated in these diseases all have roles in the repair of DNA damage, and their precise roles in maintaining telomere length are an active area of investigation. While it is currently unknown to what extent telomere erosion contributes to the normal aging process, maintenance of DNA in general and telomeric DNA, to be specific, have emerged as major players. Dr. Michael Fossel has suggested in an interview that telomerase therapies may be used not only to combat cancer but also to actually get around human aging and extend lifespan significantly. He believes human trials of telomerase-based therapies for extending lifespan will occur within the next 10 years. This timeline is significant because it coincides with the retirement of Baby Boomers in the United States and Europe.
Some experiments have raised questions on whether telomerase can be used as an anti-aging therapy, namely, the fact mice with elevated levels of telomerase have higher cancer incidence and hence do not live longer. In addition, although certain premature aging syndromes have been associated with telomere shortening, mice without active telomerase do not appear to suffer from premature aging. Telomerase also favors tumorogenesis, leading to questions about its potential as an anti-aging therapy.[15] On the other hand, one study showed that activating telomerase in cancer-resistant mice by overexpressing its catalytic subunit extended lifespan.[16] The potential remains for telomerase activators to contribute to the development of cancer.
In 2008, UCLA tested Geron's best telomerase activator named TAT2 which turned out is Cycloastragenol molecule extracted from Astragalus, according to the study.
A study in 2009, which focused on Ashkenazi Jews, found that those that live the longest inherit a hyperactive version of telomerase that rebuilds telomeres.[17]
Cancer
When cells are approaching the Hayflick limit in cell cultures, the time to senescence can be extended by the inactivation of the tumor suppressor proteins - TP53 and Retinoblastoma protein (pRb). Cells that have been so-altered will eventually undergo an event termed a "crisis" when the majority of the cells in the culture die. Sometimes, a cell does not stop dividing once it reaches crisis. In a typical situation, the telomeres are lost, and the integrity of the chromosomes declines with every subsequent cell division. Exposed chromosome ends are interpreted as double-stranded breaks (DSB) in DNA; such damage is usually repaired by reattaching (religating) the broken ends together. When the cell does this due to telomere-shortening, the ends of different chromosomes can be attached together. This temporarily solves the problem of lacking telomeres; but, during anaphase of cell division, the fused chromosomes are randomly ripped apart, causing many mutations and chromosomal abnormalities. As this process continues, the cell's genome becomes unstable. Eventually, either sufficient damage will be done to the cell's chromosomes such that cell dies (via programmed cell death, apoptosis), or an additional mutation that activates telomerase will take place.
With the activation of telomerase, some types of cells and their offspring become immortal, that is, their chromosomes will not become unstable no matter how many cell divisions they undergo (they bypass the Hayflick limit), thus avoiding cell death as long as the conditions for their duplication are met. Many cancer cells are considered 'immortal' because telomerase activity allows them to divide virtually forever, which is why they can form tumors. A good example of cancer cells' immortality is HeLa cells, which have been used in laboratories as a model cell line since 1951.
While this method of modeling human cancer in cell culture is effective and has been used for many years by scientists, it is also very imprecise. The exact changes that allow for the formation of the tumorigenic clones in the above-described experiment are not clear. Scientists have subsequently been able to address this question by the serial introduction of several mutations present in a variety of human cancers. This has led to the elucidation of several combinations of mutations that are sufficient for the formation of tumorigenic cells, in a variety of cell types. While the combination varies depending on the cell type, a common theme is that the following alterations are required: activation of TERT, loss of p53 pathway function, loss of pRb pathway function, activation of the Ras or myc proto-oncogenes, and aberration of the PP2A protein phosphatase. That is to say, the cell has an activated telomerase, eliminating the process of death by chromosome instability or loss, absence of apoptosis-induction pathways, and continued activation of mitosis.
This model of cancer in cell culture accurately describes the role of telomerase in actual human tumors. Telomerase activation has been observed in ~90% of all human tumors, suggesting that the immortality conferred by telomerase plays a key role in cancer development. Of the tumors that have not activated TERT,[18] most have found a separate pathway to maintain telomere length termed ALT (Alternative Lengthening of Telomeres).[19] The exact mechanism behind telomere maintenance in the ALT pathway has not been elucidated, but likely involves multiple recombination events at the telomere.
Additional roles in cancer, heart disease, and a socioeconomic and quality of life aspect
Additional roles for telomerase per work by Elizabeth Blackburn et al., include the upregulation of 70 genes known or suspected in cancers' growth and spread through the body, and the activation of glycolysis, which enables cancer cells to rapidly use sugar to facilitate their programmed growth rate.(roughly the growth rate of a fetus)
E. V. Gostjeva et al. (MIT) recently imaged colon cancer stem cells and compared them to fetal colon stem cells trying to make a new colon; they were the same.
Elizabeth Blackburn et al. UCSF has shown work that reveals that mothers caring for their very sick children have shorter telomeres when they report that their emotional stress is at the greatest point. She also found telomerase active at the site of blockages in coronary artery tissue. This could be why heart attacks can come on so suddenly: Telomerase is driving the growth of the blockage.
Other work has shown that the poor of society have shorter telomeres than the rich.[20] Short telomeres can lead to telomeric crisis and the initiation of cancer if many other conditions are also met.
Blackburn and the two other co-discoverers of telomerase won the Lasker Award (2006), and the Nobel Prize (2009) for the discovery of telomerase and subsequent work on telomerase. Blackburn also won the 2006 Gruber Genetics Prize for same.
Role in other human diseases
Mutations in TERT have been implicated in predisposing patients to aplastic anemia, a disorder in which the bone marrow fails to produce blood cells, in 2005.[21]
Cri du chat Syndrome (CdCS) is a complex disorder involving the loss of the distal portion of the short arm of chromosome 5. TERT is located in the deleted region, and loss of one copy of TERT has been suggested as a cause or contributing factor of this disease.[22]
Dyskeratosis congenita (DC) is a disease of the bone marrow that can be caused by some mutations in the telomerase subunits.[23] In the DC cases, about 35% cases are X-linked-recessive on the DKC1 locus[24] and 5% cases are autosomal dominant on the TERT[25] and TERC[26] loci.
Patients with DC have severe bone marrow failure manifesting as abnormal skin pigmentation, leucoplakia (a white thickening of the oral mucosa), and nail dystrophy, as well as a variety of other symptoms. Individuals with either TERC or DKC1 mutations have shorter telomeres and defective telomerase activity in vitro than other individuals of the same age.[27]
There has also been one family in which autosomal dominant DC has been linked to a heterozygous mutation in TERT.[28] These patients also exhibited an increased rate of telomere-shortening, and genetic anticipation (i.e., the DC phenotype worsened with each generation).
Telomerase as a potential drug target
Cancer is a very difficult disease to fight because the immune system has trouble recognizing it, and cancer cells are immortal; they will always continue dividing. Because telomerase is necessary for the immortality of so many cancer types, it is thought to be a potential drug target. If a drug can be used to turn off telomerase in cancer cells, the above process of telomere-shortening will resume—telomere length will be lost as the cells continue to divide, mutations will occur, and cell stability will decrease. Experimental drug and vaccine therapies targeting active telomerase have been tested in mouse models, and some have now entered early clinical trials. Geron Corporation is currently conducting four human clinical trials involving telomerase inhibition and telomerase vaccination. Merck, as a licensee of Geron, has recent approval of an IND for one vaccine type. The vaccine platform is being tested (and now jointly with Merck) using three different approaches. One vaccine is adenovirus/plasmid based (Merck IND). The second is an autologous dendritic cell based vaccine (GRNVAC1), formerly called TVAX when tested in Phase I clinical trials in Prostate Cancer, and it showed significant PSA doubling times as well as T-cell response. Geron's embryonic stem cell derived dendritic cell vaccine targeting telomerase is the third approach and is currently at the pre-clinical stage. These vaccine methods attempt to teach the human immune system to attack cancer cells expressing telomerase. Geron's telomerase inhibitor drug (GRN163L) attempts to stop cancer cell proliferation by inhibiting telomerase and it is in three separate early stage human clinical trials. Indeed, telomerase inhibition in many types of cancer cells grown in culture has led to the massive death of the cell population. However, a variety of caveats, including the presence of the ALT pathway,[18][19] complicate such therapies. Some have reported ALT methods of telomere maintenance and storage of DNA in cancer stem cells, however Geron claims to have killed cancer stem cells with their telomerase inhibitor GRN163L at Johns Hopkins. GRN163L binds directly to the RNA template of telomerase. Even a mutation of the RNA template of telomerase would render the telomerase unable to extend telomeres, and therefore not be able to grant replicative immortality to cancer, not allow glycolysis to be inititated, and not upregulate Blackburn's 70 cancer genes. Since Blackburn has shown that most of the harmful cancer-related effects of telomerase are dependent on an intact RNA template, it seems a very worthwhile target for drug development. If indeed some cancer stem cells use an alternative method of telomere maintenance, it should be noted that they are still killed when the RNA template of telomerase is blocked. According to Blackburn's opinion at most of her lectures, it is a big mistake to think that telomerase is involved with only extending telomeres. Stopping glycolysis in cancer stem cells and preventing the upregulation of 70 bad genes is probably what is killing cancer stem cells if they are using alternative methods.
See also
References
- ↑ Olovnikov AM. A theory of marginotomy. The incomplete copying of template margin in enzymatic synthesis of polynucleotides and biological significance of the phenomenon. J Theor Biol. 1973 Sep 14;41(1):181-90
- ↑ Greider, C.W. & Blackburn, E.H. (1985). "Identification of a specific telomere terminal transferase activity in Tetrahymena extracts". Cell 43 (2 Pt 1): 405–413. doi:10.1016/0092-8674(85)90170-9. PMID 3907856.
- ↑ http://nobelprize.org/nobel_prizes/medicine/laureates/2009/press.html
- ↑ Cohen S, Graham M, Lovrecz G, Bache N, Robinson P, Reddel R (2007). "Protein composition of catalytically active human telomerase from immortal cells". Science 315 (5820): 1850–3. doi:10.1126/science.1138596. PMID 17395830.
- ↑ HUGO Gene Nomenclature Committee(HGNC)- TERT
- ↑ HGNC - TERC
- ↑ HGNC - DKC1
- ↑ HGNC - TEP1
- ↑ NCBI - telomerase reverse transcriptase isoform 1
- ↑ NCBI - telomerase reverse transcriptase
- ↑ Gillis, A. J.; Schuller, A. P.; Skordalakes, E. (2008). "Structure of the Tribolium castaneum telomerase catalytic subunit TERT". Nature 455 (7213): 633–637. doi:10.1038/nature07283. PMID 18758444
- ↑ Gavory G, Farrow M, Balasubramanian S (2002). "Minimum length requirement of the alignment domain of human telomerase RNA to sustain catalytic activity in vitro". Nucleic Acids Res. 30 (20): 4470–80. doi:10.1093/nar/gkf575. PMID 12384594. PMC 137139. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC137139/?tool=pubmed.
- ↑ Hayflick L, Moorhead PS (1961). "The serial cultivation of human diploid cell strains". Exp Cell Res 25: 585–621. doi:10.1016/0014-4827(61)90192-6. PMID 13905659.
- ↑ Blasco MA. Telomeres and human disease: aging, cancer, and beyond. Nat Rev Genet. 2005 Aug;6(8):611-22. PMID 16136653
- ↑ de Magalhaes JP, Toussain O. Telomeres and telomerase: a modern fountain of youth? Rejuvenation Res. 2004 Summer;7(2):126-33 PMID 15312299
- ↑ Tomás-Loba A, Flores I, Fernández-Marcos PJ, Cayuela ML, Maraver A, Tejera A, Borrás C, Matheu A, Klatt P, Flores JM, Viña J, Serrano M, Blasco MA. Telomerase reverse transcriptase delays aging in cancer-resistant mice. Cell. 2008 Nov 14;135(4):609-22 PMID 19013273
- ↑ Britt, Robert Roy (2009). One Key Found for Living to 100. LiveScience. http://www.livescience.com/health/091112-anti-aging-telomeres.html.
- ↑ 18.0 18.1
 Bryan TM, Englezou A, Gupta J, Bacchetti S, Reddel RR. Telomere elongation in immortal human cells without detectable telomerase activity. EMBO J. 1995 Sep 1;14(17):4240-8. PMID 7556065
Bryan TM, Englezou A, Gupta J, Bacchetti S, Reddel RR. Telomere elongation in immortal human cells without detectable telomerase activity. EMBO J. 1995 Sep 1;14(17):4240-8. PMID 7556065 - ↑ 19.0 19.1 Henson JD, Neumann AA, Yeager TR, Reddel RR (2002). "Alternative lengthening of telomeres in mammalian cells". Oncogene 21 (4): 598–610. doi:10.1038/sj.onc.1205058. PMID 11850785. http://www.nature.com/onc/journal/v21/n4/full/1205058a.html.
- ↑ Reuters (2006-07-21). "The poor age faster than the rich: study". Sydney Morning Herald. http://www.smh.com.au/news/World/The-poor-age-faster-than-the-rich-study/2006/07/21/1153166552145.html
- ↑ Yamaguchi, H.; Calado, R. T.; Ly, H.; Kajigaya, S.; Baerlocher, G. M.; Chanock, S. J.; Lansdorp, P. M.; Young, N. S. (2005). "Mutations in TERT, the gene for telomerase reverse transcriptase, in aplastic anemia". New England Journal of Medicine 352 (14): 1413–1424. doi:10.1056/NEJMoa042980. PMID 15814878
- ↑ Zhang, A.; Zheng, C.; Hou, M.; Lindvall, C.; Li, K. J.; Erlandsson, F.; Bjorkholm, M.; Gruber, A. et al. (2003). "Deletion of the telomerase reverse transcriptase gene and haploinsufficiency of telomere maintenance in Cri du chat syndrome". The American Journal of Human Genetics 72 (4): 940–948. doi:10.1086/374565. PMID 12629597
- ↑ Yamaguchi H (2007). "Mutations of telomerase complex genes linked to bone marrow failures" (PDF). J Nippon Med Sch. 74 (3): 202–9. doi:10.1272/jnms.74.202. PMID 17625368. http://www.jstage.jst.go.jp/article/jnms/74/3/202/_pdf.
- ↑ Heiss NS, Knight SW, Vulliamy TJ et al. (1998). "X-linked dyskeratosis congenita is caused by mutations in a highly conserved gene with putative nucleolar functions". Nat Genet. 19 (1): 32–8. doi:10.1038/ng0598-32. PMID 9590285.
- ↑ Vulliamy TJ, Walne A, Baskaradas A et al. (2005). "Mutations in the reverse transcriptase component of telomerase (TERT) in patients with bone marrow failure". Blood Cells Mol Dis. 34 (3): 257–63. doi:10.1016/j.bcmd.2004.12.008. PMID 15885610.
- ↑ Vulliamy T, Marrone A, Goldman F et al. (2001). "The RNA component of telomerase is mutated in autosomal dominant dyskeratosis congenita". Nature 413 (6854): 432–5. doi:10.1038/35096585. PMID 11574891.
- ↑ Marrone, A.; Walne, A.; Dokal, I. (2005). "Dyskeratosis congenita: telomerase, telomeres and anticipation". Current Opinion in Genetics & Development 15 (3): 249–257. doi:10.1016/j.gde.2005.04.004. PMID 15917199
- ↑ Armanios, M.; Chen, J. L.; Chang, Y. P.; Brodsky, R. A.; Hawkins, A.; Griffin, C. A.; Eshleman, J. R.; Cohen, A. R. et al. (2005). "Haploinsufficiency of telomerase reverse transcriptase leads to anticipation in autosomal dominant dyskeratosis congenita". PNAS 102 (44): 15960–15964. doi:10.1073/pnas.0508124102. PMID 16247010
External links
- T.A. Sciences live Q&A Event on Telomere Video on Demand event
- The Telomerase Database - A Web-based tool for telomerase research.
- Three-dimensional model of telomerase at MUN
- Telomeres and Telomerase: Their Implications in Human Health and Disease on-line lecture by Elizabeth Blackburn
- MeSH Telomerase
{kind=link}
|
||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||