Porphyria
| Porphyria | |
|---|---|
| Classification and external resources | |
| ICD-10 | E80.0-E80.2 |
| ICD-9 | 277.1 |
| MedlinePlus | 001208 |
| MeSH | C17.800.849.617 |
Porphyrias are a group of inherited or acquired disorders of certain enzymes in the heme bio-synthetic pathway (also called porphyrin pathway). They are broadly classified as acute (hepatic) porphyrias and cutaneous (erythropoietic) porphyrias, based on the site of the overproduction and accumulation of the porphyrins (or their chemical precursors). They manifest with either neurological complications or skin problems (or occasionally both). A clinically induced and histologically identical condition is called pseudoporphyria. Pseudoporphyria is characterized by normal serum and urine porphyrin levels.
The term derives from the Greek πορφύρα, porphyra, meaning "purple pigment". The name is likely to have been a reference to the purple discolouration of feces and urine in patients during an attack.[1] Although original descriptions are attributed to Hippocrates, the disease was first explained biochemically by Dr Felix Hoppe-Seyler in 1874,[2] and acute porphyrias were described by the Dutch physician Prof B.J. Stokvis in 1889.[1][3]
Contents |
Signs and symptoms
Acute porphyria
The acute, or hepatic, porphyrias primarily affect the nervous system, resulting in abdominal pain, vomiting, acute neuropathy, muscle weakness, seizures, and mental disturbances, including hallucinations, depression, anxiety, and paranoia. Cardiac arrhythmias and tachycardia (fast heart rate) may develop as the autonomic nervous system is affected. Pain can be severe and can, in some cases, be both acute and chronic in nature. Constipation is frequently present, as the nervous system of the gut is affected, but diarrhea can also occur.
Given the many presentations and the relatively uncommon occurrence of porphyria, the patient may initially be suspected to have other, unrelated conditions. For instance, the polyneuropathy of acute porphyria may be mistaken for Guillain-Barré syndrome, and porphyria testing is commonly recommended in those scenarios.[4] Systemic lupus erythematosus features photosensitivity and pain attacks and shares various other symptoms with porphyria.[5]
Not all porphyrias are genetic, and patients with liver disease who develop porphyria as a result of liver dysfunction may exhibit other signs of their condition, such as jaundice.
Patients with acute porphyria (AIP, HCP, VP) are at increased risk over their life for hepatocellular carcinoma (primary liver cancer) and may require monitoring. Other typical risk factors for liver cancer need not be present.
Cutaneous porphyria
The cutaneous, or erythropoietic, porphyrias primarily affect the skin, causing photosensitivity (photodermatitis), blisters, necrosis of the skin and gums, itching, and swelling, and increased hair growth on areas such as the forehead. Often there is no abdominal pain, distinguishing it from other porphyrias.
In some forms of porphyria, accumulated heme precursors excreted in the urine may cause various changes in color, after exposure to sunlight, to a dark reddish or dark brown color. Even a purple hue or red urine may be seen.
Diagnosis
Porphyrin studies
Porphyria is diagnosed through spectroscopy and biochemical analysis of blood, urine, and stool.[6] In general, urine estimation of porphobilinogen (PBG) is the first step if acute porphyria is suspected. As a result of feedback, the decreased production of heme leads to increased production of precursors, PBG being one of the first substances in the porphyrin synthesis pathway.[7] In nearly all cases of acute porphyria syndromes, urinary PBG is markedly elevated except for the very rare ALA dehydratase deficiency or in patients with symptoms due to hereditary tyrosinemia type I. In cases of mercury- or arsenic poisoning-induced porphyria, other changes in porphyrin profiles appear, most notably elevations of uroporphyrins I & III, coproporphyrins I & III and pre-coproporphyrin.[8]
Repeat testing during an attack and subsequent attacks may be necessary in order to detect a porphyria, as levels may be normal or near-normal between attacks. The urine screening test has been known to fail in the initial stages of a severe life threatening attack of acute intermittent porphyria.
The bulk (up to 90%) of the genetic carriers of the more common, dominantly inherited acute hepatic porphyrias (acute intermittent porphyria, hereditary coproporphyria, variegate porphyria) have been noted in DNA tests to be latent for classic symptoms and may require DNA or enzyme testing. The exception to this may be latent post-puberty genetic carriers of hereditary coproporphyria.
As most porphyrias are rare conditions, general hospital labs typically do not have the expertise, technology or staff time to perform porphyria testing. In general, testing involves sending samples of blood, stool and urine to a reference laboratory.[6] All samples to detect porphyrins must be handled properly. Samples should be taken during an acute attack, otherwise a false negative result may occur. Samples must be protected from light and either refrigerated or preserved.[6]
If all the porphyrin studies are negative, one has to consider pseudoporphyria. A careful medication review often will find the inciting cause of pseudoporphyria.
Additional tests
Further diagnostic tests of affected organs may be required, such as nerve conduction studies for neuropathy or an ultrasound of the liver. Basic biochemical tests may assist in identifying liver disease, hepatocellular carcinoma, and other organ problems.
Pathogenesis
In humans, porphyrins are the main precursors of heme, an essential constituent of hemoglobin, myoglobin, catalase, peroxidase, respiratory and P450 liver cytochromes.
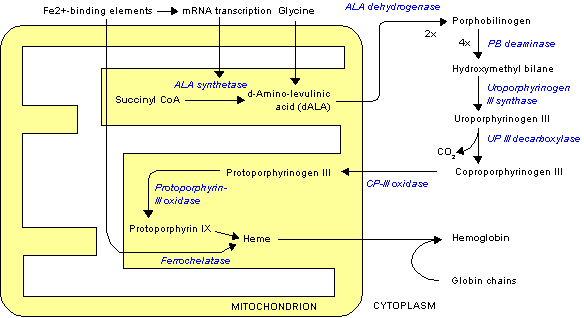
Deficiency in the enzymes of the porphyrin pathway leads to insufficient production of heme. Heme function plays a central role in cellular metabolism. This is not the main problem in the porphyrias; most heme synthesis enzymes—even dysfunctional enzymes—have enough residual activity to assist in heme biosynthesis. The principal problem in these deficiencies is the accumulation of porphyrins, the heme precursors, which are toxic to tissue in high concentrations. The chemical properties of these intermediates determine the location of accumulation, whether they induce photosensitivity, and whether the intermediate is excreted (in the urine or feces).
There are eight enzymes in the heme biosynthetic pathway, four of which—the first one and the last three—are in the mitochondria, while the other four are in the cytosol. Defects in any of these can lead to some form of porphyria.
The hepatic porphyrias are characterized by acute neurological attacks (seizures, psychosis, extreme back and abdominal pain and an acute polyneuropathy), while the erythropoietic forms present with skin problems, usually a light-sensitive blistering rash and increased hair growth.
Variegate porphyria (also porphyria variegata or mixed porphyria), which results from a partial deficiency in PROTO oxidase, manifests itself with skin lesions similar to those of porphyria cutanea tarda combined with acute neurologic attacks. All other porphyrias are either skin- or nerve-predominant.
Subtypes
Subtypes of porphyrias depend on what enzyme is deficient.
| Enzyme | Location of enzyme | Associated porphyria | Type of porphyria | Inheritance | Symptoms |
|---|---|---|---|---|---|
| δ-aminolevulinate (ALA) synthase | Mitochondrion | X-linked sideroblastic anemia (XLSA) | Erythropoietic | X-linked | |
| δ-aminolevulinate (ALA) dehydratase | Cytosol | Doss porphyria/ALA dehydratase deficiency | Hepatic | Autosomal recessive [9] | Abdominal pain, neuropathy[9] |
| hydroxymethylbilane (HMB) synthase (or PBG deaminase) | Cytosol | acute intermittent porphyria (AIP) | Hepatic | Autosomal dominant [9] | Periodic abdominal pain, peripheral neuropathy, psychiatric disorders, tachycardia[9] |
| uroporphyrinogen (URO) synthase | Cytosol | Congenital erythropoietic porphyria (CEP) | Erythropoietic | Autosomal recessive [9] | Severe photosensitivity with erythema, swelling and blistering. Hemolytic anemia, splenomegaly[9] |
| uroporphyrinogen (URO) decarboxylase | Cytosol | Porphyria cutanea tarda (PCT) | Hepatic | Autosomal dominant [9] | Photosensitivity with vesicles and bullae[9] |
| coproporphyrinogen (COPRO) oxidase | Mitochondrion | Hereditary coproporphyria (HCP) | Hepatic | Autosomal dominant [9] | Photosensitivity, neurologic symptoms, colic[9] |
| protoporphyrinogen (PROTO) oxidase | Mitochondrion | Variegate porphyria (VP) | Mixed | Autosomal dominant [9] | Photosensitivity, neurologic symptoms, developmental delay |
| Ferrochelatase | Mitochondrion | Erythropoietic protoporphyria (EPP) | Erythropoietic | Autosomal dominant [9] | Photosensitivity with skin lesions. Gallstones, mild liver dysfunction[9] |
| Transient erythroporphyria of infancy | Purpuric skin lesions[10]:526 |
Treatment
Acute porphyria
- Carbohydrates and heme
Often, empirical treatment is required if the diagnostic suspicion of a porphyria is high since acute attacks can be fatal. A high-carbohydrate diet is typically recommended; in severe attacks, a glucose 10% infusion is commenced, which may aid in recovery.
Hematin (trade name Panhematin) and heme arginate (trade name NormoSang) are the drugs of choice in acute porphyria, in the United States and the United Kingdom, respectively. These drugs need to be given very early in an attack to be effective; effectiveness varies amongst individuals. They are not curative drugs but can shorten attacks and reduce the intensity of an attack. Side effects are rare but can be serious. These heme-like substances theoretically inhibit ALA synthase and hence the accumulation of toxic precursors. In the United Kingdom, supplies of NormoSang are kept at two national centers; emergency supply is available from St Thomas' Hospital, London.[11] In the United States, Lundbeck manufactures and supplies Panhematin for infusion.[12]
Heme Arginate (NormoSang) is used during crises but also in preventive treatment to avoid crises, one treatment every 10 days
Any sign of low blood sodium (hyponatremia) or weakness should be treated with the addition of hematin or heme arginate or even Tin Mesoporphyrin as these are signs of impending syndrome of inappropriate antidiuretic hormone (SIADH) or peripheral nervous system involvement that may be localized or severe progressing to bulbar paresis and respiratory paralysis.
- Precipitating factors
If drugs or hormones have caused the attack, discontinuing the offending substances is essential. Infection is one of the top causes of attacks and requires vigorous treatment.
- Symptom control
Pain is severe, frequently out of proportion to physical signs and often requires the use of opiates to reduce it to tolerable levels. Pain should be treated early as medically possible due to its severity. Nausea can be severe; it may respond to phenothiazine drugs but is sometimes intractable. Hot water baths/showers may lessen nausea temporarily, though caution should be used to avoid burns or falls.
- Early identification
Patients with a history of acute porphyria and even genetic carriers are recommended to wear an alert bracelet or other identification at all times in case they develop severe symptoms or in case of accidents where there is a potential for drug exposure, as a result of which they are unable to explain to healthcare professionals about their condition and the fact that some drugs are absolutely contraindicated.
- Neurologic and psychiatric issues
Patients who experience frequent attacks can develop chronic neuropathic pain in extremities as well as chronic pain in the gut. Gut dysmotility, ileus, intussusception, hypoganglionosis, encopresis in children and intestinal pseudo-obstruction have been associated with porphyrias. This is thought to be due to axonal nerve deterioration in affected areas of the nervous system and vagal nerve dysfunction.
In these cases treatment with long-acting opioids may be indicated. Some cases of chronic pain can be difficult to manage and may require treatment using multiple modalities. Opioid dependence may develop.
Depression often accompanies the disease and is best dealt with by treating the offending symptoms and if needed the judicious use of anti-depressants. Some psychotropic drugs are porphyrinogenic, limiting the pharmacotherapeutic scope.
- Seizures
Seizures often accompany this disease. Most seizure medications exacerbate this condition. Treatment can be problematic: barbiturates especially must be avoided. Some benzodiazepines are safe and, when used in conjunction with newer anti-seizure medications such as gabapentin, offer a possible regime for seizure control.
Magnesium sulfate and bromides have also been used in porphyria seizures, however, development of status epilepticus in porphyria may not respond to magnesium alone. The addition of hematin or heme arginate has been used during status epilepticus.
- Underlying liver disease
Some liver diseases may cause porphyria even in the absence of genetic predisposition. These include hemochromatosis and hepatitis C. Treatment of iron overload may be required.
- Hormone treatment
Hormonal fluctuations that contribute to cyclical attacks in women have been treated with oral contraceptives and luteinizing hormones to shut down menstrual cycles. However, oral contraceptives have also triggered photosensitivity and withdrawal of oral contraceptives has triggered attacks. Androgens and fertility hormones have also triggered attacks.
Erythropoietic porphyrias
These are associated with accumulation of porphyrins in erythrocytes and are rare. The rarest is Congenital erythropoetic porphyria (C.E.P) otherwise known as Gunther's disease. The signs may present from birth and include severe photosensitivity, brown teeth that fluoresce in ultraviolet light due to deposition of type one porphyrins and later hypertrichosis. Hemolytic anemia usually develops. Pharmaceutical-grade beta carotene may be used in its treatment.[13] A bone marrow transplant has also been successful in curing CEP in a few cases, although long term results are not yet available.[14]
The pain, burning, swelling and itching that occur in erythropoietic porphyrias generally require avoidance of bright sunlight. Most kinds of sunscreen are not effective, but SPF-rated long-sleeve shirts, hats, bandanas and gloves can help. Chloroquine may be used to increase porphyrin secretion in some EPs.[6] Blood transfusion is occasionally used to suppress innate heme production.
Culture and history
Porphyrias have been detected in all races, multiple ethnic groups on every continent including Africans, Asians, Australian aborigines, Caucasians, Peruvian, Mexican, Native Americans, and Sami. There are high incidence reports of AIP in areas of India and Scandinavia and over 200 genetic variants of AIP, some of which are specific to families, although some strains have proven to be repeated mutations.
The links between porphyrias and mental illness have been noted for decades. In the early 1950s patients with porphyrias (occasionally referred to as "Porphyric Hemophilia"[15]) and severe symptoms of depression or catatonia were treated with electroshock.
Vampires and werewolves
Porphyria has been suggested as an explanation for the origin of vampire and werewolf legends, based upon certain perceived similarities between the condition and the folklore.
In January 1964, L. Illis' 1963 paper, "On Porphyria and the Aetiology of Werwolves", was published in Proceedings of the Royal Society of Medicine. Later, Nancy Garden argued for a connection between porphyria and the vampire belief in her 1973 book, Vampires. In 1985, biochemist David Dolphin's paper for the American Association for the Advancement of Science, "Porphyria, Vampires, and Werewolves: The Aetiology of European Metamorphosis Legends", gained widespread media coverage, thus popularizing the connection.
The theory has since faced heavy criticism, especially for the stigma it has placed on its sufferers. Norine Dresser's American Vampires: Fans, Victims, Practitioners (1989) treats the matter with more depth.
The theory also operates on a highly-flawed premise, mainly in regard to a perceived harmful effect sunlight had on vampires, a property relatively late to vampire belief. There are about eight different types of porphyria, four of these types of porphyria can sometimes cause sensitivity to light: Erythropoietic Protoporphyria (EPP) or Protoporphyria, Congenital Erythropoietic Porphyria (C.E.P.), Porphyria Cutanea Tarda (PCT) and Variegate Porphyria.
Notable cases

The mental illness exhibited by King George III evidenced in the regency crisis of 1788 has inspired several attempts at retrospective diagnosis. The first, written in 1855, thirty-five years after his death, concluded he suffered from acute mania. M. Guttmacher, in 1941, suggested manic-depressive psychosis as a more likely diagnosis. The first suggestion that a physical illness was the cause of King George's mental derangements came in 1966, in a paper "The Insanity of King George III: A Classic Case of Porphyria",[16] with a follow-up in 1968, "Porphyria in the Royal Houses of Stuart, Hanover and Prussia".[17] The papers, by a mother/son psychiatrist team, were written as though the case for porphyria had been proven, but the response demonstrated that many, including those more intimately familiar with actual manifestations of porphyria, were unconvinced. Many psychiatrists disagreed with Hunter's diagnosis, suggesting bipolar disorder as far more probable. The theory is treated in Purple Secret,[18] which documents the ultimately unsuccessful search for genetic evidence of porphyria in the remains of royals suspected to suffer from it.[19] In 2005 it was suggested that arsenic (which is known to be porphyrogenic) given to George III with antimony may have caused his porphyria.[20] Despite the lack of direct evidence, the notion that George III (and other members of the royal family) suffered from porphyria has achieved such popularity that many forget that it is merely a hypothesis. In 2010 an exhaustive analysis of historical records revealed that the porphyria claim was based on spurious and selective interpretation of contemporary medical and historical sources.[21]
The mental illness of George III is the basis of the plot in The Madness of King George, a 1994 British film based upon the 1991 Alan Bennett play, The Madness of George III. The closing credits of the film include the comment that the illness suffered by King George has been attributed to porphyria and that it is hereditary. Among other descendants of George III theorised by the authors of Purple Secret to suffering from porphyria (based upon analysis of their extensive and detailed medical correspondence) were his great-great-granddaughter Princess Charlotte of Prussia (Emperor William II's eldest sister) and her daughter Princess Feodora of Saxe-Meiningen. They had more success in being able to uncover reliable evidence that George III's great-great-great-grandson Prince William of Gloucester was reliably diagnosed with variegate porphyria.

It is believed that Mary, Queen of Scots – King George III's great-great-great-great-great-grandmother – also suffered from acute intermittent porphyria, although this is subject to much debate. It is assumed she inherited the disorder, if indeed she had it, from her father, James V of Scotland; both father and daughter endured well-documented attacks that some believe fall within the constellation of symptoms of porphyria.
Vlad III the Impaler was also said to have suffered from Acute Porphyria, which may have started the notion that Vampires were, "allergic to the light of day."
Other commentators have suggested that Vincent van Gogh may have suffered from acute intermittent porphyria.[22] It has also been imagined that King Nebuchadnezzar of Babylon suffered from some form of porphyria (cf. Daniel 4).[23] However, the symptoms of the various porphyrias are so extensive that a wide constellation of symptoms can be attributed to one or more of them.
Paula Frías Allende, the daughter of the Chilean novelist Isabel Allende, fell into a porphyria-induced coma in 1991 which inspired Isabel Allende to write the autobiographical book Paula, dedicated to her daughter.
Julia Gnuse, regarded as the most tattooed woman in the world, got hers to hide scars from porphyria cutanea tarda (PCT).
References
- ↑ 1.0 1.1 Nick Lane (2002-12-16). "Born to the purple: the story of porphyria". Scientific American. http://www.sciam.com/article.cfm?articleID=000B1BEF-C051-1DF8-9733809EC588EEDF. Retrieved 2008-08-05.
- ↑ Hoppe-Seyler F (1871). "Das Hämatin". Tubinger Med-Chem Untersuch 4: 523–33.
- ↑ Stokvis BJ. "Over twee zeldzame kleurstoffen in urine van zieken" (in Dutch). Nederl Tijdschr Geneeskd 2: 409–417. Reprinted in Stokvis BJ (December 1989). "Over twee zeldzame kleurstoffen in urine van zieken" (in Dutch). Ned Tijdschr Geneeskd 133 (51): 2562–70. PMID 2689889.
- ↑ Albers JW, Fink JK (2004). "Porphyric neuropathy". Muscle Nerve 30 (4): 410–22. doi:10.1002/mus.20137. PMID 15372536.
- ↑ Roelandts R (2000). "The diagnosis of photosensitivity". Arch Dermatol 136 (9): 1152–7. doi:10.1001/archderm.136.9.1152. PMID 10987875.
- ↑ 6.0 6.1 6.2 6.3 Thadani H, Deacon A, Peters T (2000). "Diagnosis and management of porphyria". BMJ 320 (7250): 1647–51. doi:10.1136/bmj.320.7250.1647. PMID 10856069.
- ↑ Anderson KE, Bloomer JR, Bonkovsky HL, et al. (2005). "Recommendations for the diagnosis and treatment of the acute porphyrias". Ann. Intern. Med. 142 (6): 439–50. PMID 15767622.
- ↑ Woods, J.S. (1995). "Porphyrin metabolism as indicator of metal exposure and toxicity". In Goyer, R.A. & Cherian, M.G.. Toxicology of metals, biochemical aspects. 115. Berlin: Springer. pp. 19–52, Chapter 2.
- ↑ 9.00 9.01 9.02 9.03 9.04 9.05 9.06 9.07 9.08 9.09 9.10 9.11 9.12 Table 18-1 in: Marks, Dawn B.; Swanson, Todd; Sandra I Kim; Marc Glucksman (2007). Biochemistry and molecular biology. Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins. ISBN 0-7817-8624-X.
- ↑ James, William D.; Berger, Timothy G.; et al. (2006). Andrews' Diseases of the Skin: clinical Dermatology. Saunders Elsevier. ISBN 0-7216-2921-0.
- ↑ "9.8.2: Acute porphyrias". British National Formulary (BNF 57). United Kingdom: BMJ Group and RPS Publishing. March 2009. p. 549. ISBN 9780853698456.
- ↑ American Porphyria Foundation (2010). "Panhematin for Acute Porphyria". http://www.porphyriafoundation.com/testing-and-treatment/medications-for-porphyria/panhematin. Retrieved 2010-08-05.
- ↑ Martin A Crook.2006. Clinical chemistry and Metabolic Medicine. seventh edition. Hodder Arnold. ISBN 0-340-90616-2
- ↑ Faraci M, Morreale G, Boeri E, et al. (2008). "Unrelated HSCT in an adolescent affected by congenital erythropoietic porphyria". Pediatr Transplant 12 (1): 117–20. doi:10.1111/j.1399-3046.2007.00842.x. PMID 18186900.
- ↑ Denver, Joness. "An Encyclopaedia of Obscure Medicine". Published by University Books, Inc., 1959.
- ↑ Macalpine I, Hunter R (January 1966). "The "insanity" of King George 3d: a classic case of porphyria". Br Med J 1 (5479): 65–71. doi:10.1136/bmj.1.5479.65. PMID 5323262.
- ↑ Macalpine I, Hunter R, Rimington C (January 1968). "Porphyria in the royal houses of Stuart, Hanover, and Prussia. A follow-up study of George 3d's illness". Br Med J 1 (5583): 7–18. doi:10.1136/bmj.1.5583.7. PMID 4866084.
- ↑ Warren, Martin; Rh̲l, John C. G.; Hunt, David C. (1998). Purple secret: genes, "madness" and the Royal houses of Europe. London: Bantam. ISBN 0-593-04148-8.
- ↑ The authors demonstrated a single point mutation in the PPOX gene, but not one which has been associated with disease.
- ↑ Cox TM, Jack N, Lofthouse S, Watling J, Haines J, Warren MJ (2005). "King George III and porphyria: an elemental hypothesis and investigation". Lancet 366 (9482): 332–5. doi:10.1016/S0140-6736(05)66991-7. PMID 16039338.
- ↑ Peters, Timothy J. and Wilkinson, D. (2010) "King George III and porphyria: a clinical re-examination of the historical evidence", History of Psychiatry 21: pp 3–19.
- ↑ Loftus LS, Arnold WN (1991). "Vincent van Gogh's illness: acute intermittent porphyria?". BMJ 303 (6817): 1589–91. doi:10.1136/bmj.303.6817.1589. PMID 1773180.
- ↑ Beveridge A (2003). "The madness of politics". J R Soc Med 96 (12): 602–4. doi:10.1258/jrsm.96.12.602. PMID 14645615.
External links
- American Porphyria Foundation
- European Porphyria Initiative
- The British Porphyria Association
- The Drug Database for Acute Porphyria - comprehensive database on drug porphyrinogenicity
|
|||||||||||||||||||||||