Phylogenetics
| Part of the Biology series on |
| Evolution |
| Mechanisms and processes |
|---|
|
Adaptation |
| Research and history |
|
Introduction |
| Evolutionary biology fields |
|
Cladistics |
| Biology portal · |
In biology, phylogenetics is the study of evolutionary relatedness among various groups of organisms (for example, species or populations), which is discovered through molecular sequencing data and morphological data matrices. The term phylogenetics is of Greek origin from the terms phyle/phylon (φυλή/φῦλον), meaning "tribe, race," and genetikos (γενετικός), meaning "relative to birth" from genesis (γένεσις, "birth"). Taxonomy, the classification, identification, and naming of organisms, has been richly informed by phylogenetics but remains methodologically and logically distinct.[1] The fields overlap however in the science of phylogenetic systematics – often called "cladism" or "cladistics" –, where only phylogenetic trees are used to delimit taxa, which represent groups of lineage-connected individuals.[2] In biological systematics as a whole, phylogenetic analyses have become essential in researching the evolutionary tree of life.
Contents |
Construction of a phylogenetic tree
Evolution is regarded as a branching process, whereby populations are altered over time and may speciate into separate branches, hybridize together, or terminate by extinction. This may be visualized in a phylogenetic tree.
The problem posed by phylogenetics is that genetic data are only available for the present, and fossil records (osteometric data) are sporadic and less reliable. Our knowledge of how evolution operates is used to reconstruct the full tree.[3] Thus, a phylogenetic tree is based on a hypothesis of the order in which evolutionary events are assumed to have occurred.
Cladistics is the current method of choice to infer phylogenetic trees. The most commonly-used methods to infer phylogenies include parsimony, maximum likelihood, and MCMC-based Bayesian inference. Phenetics, popular in the mid-20th century but now largely obsolete, uses distance matrix-based methods to construct trees based on overall similarity, which is often assumed to approximate phylogenetic relationships. All methods depend upon an implicit or explicit mathematical model describing the evolution of characters observed in the species included, and are usually used for molecular phylogeny, wherein the characters are aligned nucleotide or amino acid sequences.
Grouping of organisms
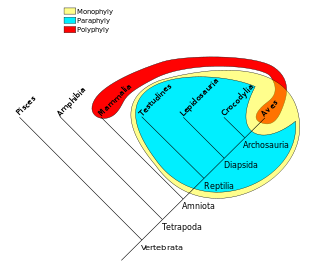
There are some terms that describe the nature of a grouping in such trees. For instance, all birds and reptiles are believed to have descended from a single common ancestor, so this taxonomic grouping (yellow in the diagram below) is called monophyletic. "Modern reptile" (cyan in the diagram) is a grouping that contains a common ancestor, but does not contain all descendants of that ancestor (birds are excluded). This is an example of a paraphyletic group. A grouping such as warm-blooded animals would include only mammals and birds (red/orange in the diagram) and is called polyphyletic because the members of this grouping do not include the most recent common ancestor.
Molecular phylogenetics
The evolutionary connections between organisms are represented graphically through phylogenetic trees. Due to the fact that evolution takes place over long periods of time that cannot be observed directly, biologists must reconstruct phylogenies by inferring the evolutionary relationships among present-day organisms. Fossils can aid with the reconstruction of phylogenies; however, fossil records are often too poor to be of good help. Therefore, biologists tend to be restricted with analysing present-day organisms to identify their evolutionary relationships. Phylogenetic relationships in the past were reconstructed by looking at phenotypes, often anatomical characteristics. Today, molecular data, which includes protein and DNA sequences, are used to construct phylogenetic trees.[4] The overall goal of National Science Foundation's Assembling the Tree of Life activity (AToL) is to resolve evolutionary relationships for large groups of organisms throughout the history of life, with the research often involving large teams working across institutions and disciplines. Investigators are typically supported for projects in data acquisition, analysis, algorithm development and dissemination in computational phylogenetics and phyloinformatics. For example, RedToL aims at reconstructing the Red Algal Tree of Life.
Ernst Haeckel's recapitulation theory

During the late 19th century, Ernst Haeckel's recapitulation theory, or biogenetic law, was widely accepted. This theory was often expressed as "ontogeny recapitulates phylogeny", i.e. the development of an organism exactly mirrors the evolutionary development of the species. Haeckel's early version of this hypothesis [that the embryo mirrors adult evolutionary ancestors] has since been rejected, and the hypothesis amended as the embryo's development mirroring embryos of its evolutionary ancestors. He was accused by five professors of falsifying his images of embryos (See Ernst Haeckel). Most modern biologists recognize numerous connections between ontogeny and phylogeny, explain them using evolutionary theory, or view them as supporting evidence for that theory. Donald I. Williamson suggested that larvae and embryos represented adults in other taxa that have been transferred by hybridization (the larval transfer theory).[5][6] However, Williamson's views do not represent mainstream thought in molecular biology[7], and there is a significant body of evidence against the larval transfer theory.[8]
Gene transfer
In general, organisms can inherit genes in two ways: vertical gene transfer and horizontal gene transfer. Vertical gene transfer is the passage of genes from parent to offspring, and horizontal gene transfer or lateral gene transfer occurs when genes jump between unrelated organisms, a common phenomenon in prokaryotes.
Horizontal gene transfer has complicated the determination of phylogenies of organisms, and inconsistencies in phylogeny have been reported among specific groups of organisms depending on the genes used to construct evolutionary trees.
Carl Woese came up with the three-domain theory of life (eubacteria, archaea and eukaryotes) based on his discovery that the genes encoding ribosomal RNA are ancient and distributed over all lineages of life with little or no horizontal gene transfer. Therefore, rRNAs are commonly recommended as molecular clocks for reconstructing phylogenies.
This has been particularly useful for the phylogeny of microorganisms, to which the species concept does not apply and which are too morphologically simple to be classified based on phenotypic traits.
Taxon sampling and phylogenetic signal
Owing to the development of advanced sequencing techniques in molecular biology, it has become feasible to gather large amounts of data (DNA or amino acid sequences) to infer phylogenetic hypotheses. For example, it is not rare to find studies with character matrices based on whole mitochondrial genomes (~16,000 nucleotides, in many animals). However, it has been proposed that it is more important to increase the number of taxa in the matrix than to increase the number of characters, because the more taxa the more robust is the resulting phylogenetic tree [9] . This may be partly due to the breaking up of long branches. It has been argued that this is an important reason to incorporate data from fossils into phylogenies where possible. Of course, phylogenetic data that include fossil taxa are generally based on morphology, rather than DNA data. Using simulations, Derrick Zwickl and David Hillis[10] found that increasing taxon sampling in phylogenetic inference has a positive effect on the accuracy of phylogenetic analyses.
Another important factor that affects the accuracy of tree reconstruction is whether the data analyzed actually contain a useful phylogenetic signal, a term that is used generally to denote whether related organisms tend to resemble each other with respect to their genetic material or phenotypic traits.[11] Ultimately, however, there is no way to measure whether a particular phylogenetic hypothesis is accurate or not, unless the "true" relationships among the taxa being examined are already known. The best result an empirical systematist can hope to attain is a tree with branches well-supported by the available evidence.
See also
- Bauplan
- Bioinformatics
- Biomathematics
- Cladistics
- Coalescent theory
- Computational phylogenetics
- EDGE of Existence Programme
- Important publications in phylogenetics
- Language family
- Maximum parsimony
- Molecular phylogeny
- PhyloCode
- Joe Felsenstein
- Systematics
- Phylogenetic tree
- Phylogenetic network
- Phylogenetic nomenclature
- Phylogenetics software
- Phylogenetic tree viewers
- Phylogeography
- Phylodynamics
- Phylogenetic comparative methods
- Microbial phylogenetics
References
- ↑ Edwards AWF, Cavalli-Sforza LL Phylogenetics is that branch of life science,which deals with the study of evolutionary relation among various groups of organisms,through molecular sequencing data. (1964). Systematics Assoc. Publ. No. 6: Phenetic and Phylogenetic Classification. ed. Reconstruction of evolutionary trees. pp. 67–76.
- ↑ Speer, Vrian (1998). "UCMP Glossary: Phylogenetics". UC Berkeley. http://www.ucmp.berkeley.edu/glossary/glossary_1.html. Retrieved 2008-03-22.
- ↑ Cavalli-Sforza LL, Edwards AWF (Sep., 1967). "Phylogenetic analysis: Models and estimation procedures". Evol. 21 (3): 550–570. doi:10.2307/2406616. http://links.jstor.org/sici?sici=0014-3820%28196709%2921%3A3%3C550%3APAMAEP%3E2.0.CO%3B2-I.
- ↑ Pierce, Benjamin A. (2007-12-17). Genetics: A conceptual Approach (3rd ed.). W. H. Freeman. ISBN 978-0716-77928-5.
- ↑ Williamson DI (2003-12-31). "xviii". The Origins of Larvae (2nd ed.). Springer. pp. 261. ISBN 978-1402-01514-4.
- ↑ Williamson DI (2006). "Hybridization in the evolution of animal form and life-cycle". Zoological Journal of the Linnean Society 148: 585–602. doi:10.1111/j.1096-3642.2006.00236.x.
- ↑ John Timmer, "Examining science on the fringes: vital, but generally wrong", ARS Technica, 9 November 2009
- ↑ Michael W. Hart, and Richard K. Grosberg, "Caterpillars did not evolve from onychophorans by hybridogenesis", Proceedings of the National Academy of the Sciences, 30 October 2009 (doi: 10.1073/pnas.0910229106)
- ↑ Wiens J (2006). "Missing data and the design of phylogenetic analyses". Journal of Biomedical Informatics 39 (1): 34–42. doi:10.1016/j.jbi.2005.04.001. PMID 15922672.
- ↑ Zwickl DJ, Hillis DM (2002). "Increased taxon sampling greatly reduces phylogenetic error". Systematic Biology 51 (4): 588–598. doi:10.1080/10635150290102339. PMID 12228001.
- ↑ Blomberg SP, Garland T Jr, Ives AR (2003). "Testing for phylogenetic signal in comparative data: behavioral traits are more labile". Evolution 57 (4): 717–745. PMID 12778543. PDF
Further reading
- Schuh, R. T. and A. V. Z. Brower. 2009. Biological Systematics: principles and applications (2nd edn.) ISBN 978-0-8014-4799-0
External links
- The Tree of Life
- Interactive Tree of Life
- PhyloCode
- ExploreTree
- UCMP Exhibit Halls: Phylogeny Wing
- Willi Hennig Society
- Filogenetica.org in Spanish
- PhyloPat, Phylogenetic Patterns
- SplitsTree, program for computing phylogenetic trees and unrooted phylogenetic networks
- Dendroscope, program for drawing phylogenetic trees and rooted phylogenetic networks
- Phylogenetic inferring on the T-REX server
- Mesquite
- Geneious Pro all-in-one phylogenetics software
- NCBI - Systematics and Molecular Phylogenetics
- What Genomes Can Tell Us About the Past - lecture on phylogenetics by Sydney Brenner
- Mikko's Phylogeny Archive
- Phylogenetic Reconstruction from Gene-Order Data
|
||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||