HIV
| HIV | |
|---|---|
| Classification and external resources | |
 The Red ribbon is a symbol for solidarity with HIV-positive people and those living with AIDS. |
|
| ICD-10 | B20-B24 |
| ICD-9 | 042-044 |
| OMIM | 609423 |
| MedlinePlus | 000602 |
| eMedicine | article/783434 |
| MeSH | D006678 |
| Human immunodeficiency virus | |
|---|---|
 |
|
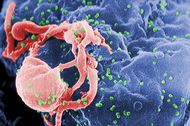
| Scanning electron micrograph of HIV-1 (in green) budding from cultured lymphocyte. Multiple round bumps on cell surface represent sites of assembly and budding of virions. | |
| Virus classification | |
| Group: | Group VI (ssRNA-RT) |
| Family: | Retroviridae |
| Genus: | Lentivirus |
| Species | |
|
|
Human immunodeficiency virus (HIV) is a lentivirus (a member of the retrovirus family) that causes acquired immunodeficiency syndrome (AIDS),[1][2] a condition in humans in which the immune system begins to fail, leading to life-threatening opportunistic infections. Infection with HIV occurs by the transfer of blood, semen, vaginal fluid, pre-ejaculate, or breast milk. Within these bodily fluids, HIV is present as both free virus particles and virus within infected immune cells. The four major routes of transmission are unsafe sex, contaminated needles, breast milk, and transmission from an infected mother to her baby at birth (perinatal transmission). Screening of blood products for HIV has largely eliminated transmission through blood transfusions or infected blood products in the developed world.
HIV infection in humans is considered pandemic by the World Health Organization (WHO). Nevertheless, complacency about HIV may play a key role in HIV risk.[3][4] From its discovery in 1981 to 2006, AIDS killed more than 25 million people.[5] HIV infects about 0.6% of the world's population.[5] In 2005 alone, AIDS claimed an estimated 2.4–3.3 million lives, of which more than 570,000 were children. A third of these deaths are occurring in Sub-Saharan Africa, retarding economic growth and increasing poverty.[6] According to current estimates, HIV is set to infect 90 million people in Africa, resulting in a minimum estimate of 18 million orphans.[7] Antiretroviral treatment reduces both the mortality and the morbidity of HIV infection, but routine access to antiretroviral medication is not available in all countries.[8]
HIV infects primarily vital cells in the human immune system such as helper T cells (to be specific, CD4+ T cells), macrophages, and dendritic cells.[9] HIV infection leads to low levels of CD4+ T cells through three main mechanisms: First, direct viral killing of infected cells; second, increased rates of apoptosis in infected cells; and third, killing of infected CD4+ T cells by CD8 cytotoxic lymphocytes that recognize infected cells. When CD4+ T cell numbers decline below a critical level, cell-mediated immunity is lost, and the body becomes progressively more susceptible to opportunistic infections.
Most untreated people infected with HIV-1 eventually develop AIDS.[10] These individuals mostly die from opportunistic infections or malignancies associated with the progressive failure of the immune system.[11] HIV progresses to AIDS at a variable rate affected by viral, host, and environmental factors; most will progress to AIDS within 10 years of HIV infection: some will have progressed much sooner, and some will take much longer.[12][13] Treatment with anti-retrovirals increases the life expectancy of people infected with HIV. Even after HIV has progressed to diagnosable AIDS, the average survival time with antiretroviral therapy was estimated to be more than 5 years as of 2005.[14] Without antiretroviral therapy, someone who has AIDS typically dies within a year.[15]
Contents |
Classification
HIV is a member of the genus Lentivirus,[16] part of the family of Retroviridae.[17] Lentiviruses have many common morphologies and biological properties. Many species are infected by lentiviruses, which are characteristically responsible for long-duration illnesses with a long incubation period.[18] Lentiviruses are transmitted as single-stranded, positive-sense, enveloped RNA viruses. Upon entry of the target cell, the viral RNA genome is converted to double-stranded DNA by a virally encoded reverse transcriptase that is present in the virus particle. This viral DNA is then integrated into the cellular DNA by a virally encoded integrase, along with host cellular co-factors,[19] so that the genome can be transcribed. After the virus has infected the cell, two pathways are possible: either the virus becomes latent and the infected cell continues to function or the virus becomes active and replicates, and a large number of virus particles that can then infect other cells are liberated.
There are two species of HIV known to exist: HIV-1 and HIV-2. HIV-1 is the virus that was initially discovered and termed both LAV and HTLV-III. It is more virulent, more infective,[20] and is the cause of the majority of HIV infections globally. The lower infectivity of HIV-2 compared to HIV-1 implies that fewer of those exposed to HIV-2 will be infected per exposure. Because of its relatively poor capacity for transmission, HIV-2 is largely confined to West Africa.[21]
| Species | Virulence | Infectivity | Prevalence | Inferred origin |
|---|---|---|---|---|
| HIV-1 | High | High | Global | Common Chimpanzee |
| HIV-2 | Lower | Low | West Africa | Sooty Mangabey |
Signs and symptoms
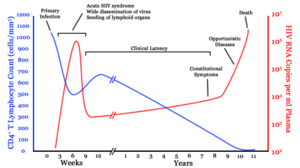
Infection with HIV-1 is associated with a progressive decrease of the CD4+ T cell count and an increase in viral load. The stage of infection can be determined by measuring the patient's CD4+ T cell count, and the level of HIV in the blood.
HIV infection has four basic stages: incubation period, acute infection, latency stage and AIDS. The initial incubation period upon infection is asymptomatic and usually lasts between two and four weeks. The second stage, acute infection, lasts an average of 28 days and can include symptoms such as fever, lymphadenopathy (swollen lymph nodes), pharyngitis (sore throat), rash, myalgia (muscle pain), malaise, and mouth and esophageal sores.
The latency stage, which occurs third, shows few or no symptoms and can last anywhere from two weeks to twenty years and beyond. AIDS, the fourth and final stage of HIV infection shows as symptoms of various opportunistic infections.
A study of French hospital patients found that approximately 0.5% of HIV-1 infected individuals retain high levels of CD4 T-cells and a low or clinically undetectable viral load without anti-retroviral treatment. These individuals are classified as HIV controllers or long-term nonprogressors.[22]
Acute HIV infection

The initial infection with HIV generally occurs after transfer of body fluids from an infected person to an uninfected one. The first stage of infection, the primary, or acute infection, is a period of rapid viral replication that immediately follows the individual's exposure to HIV leading to an abundance of virus in the peripheral blood with levels of HIV commonly approaching several million viruses per mL.[23]
This response is accompanied by a marked drop in the numbers of circulating CD4+ T cells. This acute viremia is associated in virtually all patients with the activation of CD8+ T cells, which kill HIV-infected cells, and subsequently with antibody production, or seroconversion. The CD8+ T cell response is thought to be important in controlling virus levels, which peak and then decline, as the CD4+ T cell counts rebound. A good CD8+ T cell response has been linked to slower disease progression and a better prognosis, though it does not eliminate the virus.[24]
During this period (usually 2–4 weeks post-exposure) most individuals (80 to 90%) develop an influenza or mononucleosis-like illness called acute HIV infection, the most common symptoms of which may include fever, lymphadenopathy, pharyngitis, rash, myalgia, malaise, mouth and esophageal sores, and may also include, but less commonly, headache, nausea and vomiting, enlarged liver/spleen, weight loss, thrush, and neurological symptoms. Infected individuals may experience all, some, or none of these symptoms. The duration of symptoms varies, averaging 28 days and usually lasting at least a week.[25]
Because of the nonspecific nature of these symptoms, they are often not recognized as signs of HIV infection. Even if patients go to their doctors or a hospital, they will often be misdiagnosed as having one of the more common infectious diseases with the same symptoms. As a consequence, these primary symptoms are not used to diagnose HIV infection, as they do not develop in all cases and because many are caused by other more common diseases. However, recognizing the syndrome can be important because the patient is much more infectious during this period.[26]
Latency stage
A strong immune defense reduces the number of viral particles in the blood stream, marking the start of the infection's clinical latency stage. Clinical latency can vary between two weeks and 20 years. During this early phase of infection, HIV is active within lymphoid organs, where large amounts of virus become trapped in the follicular dendritic cells (FDC) network.[27]
The surrounding tissues that are rich in CD4+ T cells may also become infected, and viral particles accumulate both in infected cells and as free virus. Individuals who are in this phase are still infectious. During this time, CD4+ CD45RO+ T cells carry most of the proviral load.[28]
AIDS
- For more details on this topic, see AIDS Diagnosis, AIDS Symptoms and WHO Disease Staging System for HIV Infection and Disease
When CD4+ T cell numbers decline below a critical level of 200 cells per µL, cell-mediated immunity is lost, and infections with a variety of opportunistic microbes appear. The first symptoms often include moderate and unexplained weight loss, recurring respiratory tract infections (such as sinusitis, bronchitis, otitis media, pharyngitis), prostatitis, skin rashes, and oral ulcerations.
Common opportunistic infections and tumors, most of which are normally controlled by robust CD4+ T cell-mediated immunity then start to affect the patient. Typically, resistance is lost early on to oral Candida species and to Mycobacterium tuberculosis, which leads to an increased susceptibility to oral candidiasis (thrush) and tuberculosis. Later, reactivation of latent herpes viruses may cause worsening recurrences of herpes simplex eruptions, shingles, Epstein-Barr virus-induced B-cell lymphomas, or Kaposi's sarcoma.
Pneumonia caused by the fungus Pneumocystis jirovecii is common and often fatal. In the final stages of AIDS, infection with cytomegalovirus (another herpes virus) or Mycobacterium avium complex is more prominent. Not all patients with AIDS get all these infections or tumors, and there are other tumors and infections that are less prominent but still significant.
Pathophysiology
Transmission
| Exposure Route | Estimated infections per 10,000 exposures to an infected source |
|||
|---|---|---|---|---|
| Blood transfusion | 9,000[31] | |||
| Childbirth | 2,500[32] | |||
| Needle-sharing injection drug use | 67[33] | |||
| Percutaneous needle stick | 30[34] | |||
| Receptive anal intercourse (2009 and 2010 studies) | 170‡ [30-890][35] / 143 [48-285][30] | |||
| Receptive anal intercourse (based on data of a 1992 study) | 50[36][37] | |||
| Insertive anal intercourse for uncircumcised men (2010 study) | 62a [7-168][30] | |||
| Insertive anal intercourse for circumcised men (2010 study) | 11a [2-24][30] | |||
| Insertive anal intercourse (based on data of a 1992 study) | 6.5[36][37] | |||
| Low-income country female-to-male | 38‡ [13-110][35] | |||
| Low-income country male-to-female | 30‡ [14-63][35] | |||
| Receptive penile-vaginal intercourse | 10[36][37][38] | |||
| Insertive penile-vaginal intercourse | 5[36][37] | |||
| Receptive oral intercourse (performed on a man) | 1†b[37] | |||
| Insertive oral intercourse (by a man) | 0.5†b[37] | |||
|
The data shown represents the rate of transmission when condoms were not used. Note that risk rates may change due to other factors such as commercial sex exposure, phase of HIV infection, presence or history of genital ulcers, and national income levels.[35] |
||||
| Bracketed values represent 95% confidence interval † "best-guess estimate" ‡ Pooled transmission probability estimate |
||||
| a Other studies found insufficient evidence that male circumcision protects against HIV infection among men who have sex with men[39][40] | ||||
| b Oral trauma, sores, inflammation, concomitant sexually transmitted infections, ejaculation in the mouth, and systemic immune suppression may increase HIV transmission rate.[41] | ||||
Three main transmission routes for HIV have been identified. HIV-2 is transmitted much less frequently by the mother-to-child and sexual route than HIV-1.
Sexual
The majority of HIV infections are acquired through unprotected sexual relations. Complacency about HIV plays a key role in HIV risk.[3][4] Sexual transmission can occur when infected sexual secretions of one partner come into contact with the genital, oral, or rectal mucous membranes of another. In high-income countries, the risk of female-to-male transmission is 0.04% per act and male-to-female transmission is 0.08% per act. For various reasons, these rates are 4 to 10 times higher in low-income countries.[35] The rate for receptive anal intercourse is much higher, 1.7% per act.[35]
The correct and consistent use of latex condoms reduces the risk of sexual transmission of HIV by about 85%.[42] However, spermicide may actually increase the transmission rate.[43][44][45]
Randomized, controlled trials in which uncircumcised men were randomly assigned to be medically circumcised in sterile conditions and given counseling and other men were not circumcised, have been conducted in South Africa,[46] Kenya,[47] and Uganda[48] showing reductions in female-to-male sexual HIV transmission of 60%, 53%, and 51% respectively. As a result, a panel of experts convened by WHO and the UNAIDS Secretariat has "recommended that male circumcision now be recognized as an additional important intervention to reduce the risk of heterosexually acquired HIV infection in men."[49] Among men who have sex with men, there is insufficient evidence that male circumcision protects against HIV infection or other Sexually Transmitted Infections.[39]
Studies of HIV among women who have undergone female genital cutting (FGC) have reported mixed results; for details see Female genital cutting#HIV.
Blood or blood product
In general, if infected blood comes into contact with any open wound, HIV may be transmitted. This transmission route can account for infections in intravenous drug users, hemophiliacs, and recipients of blood transfusions (though most transfusions are checked for HIV in the developed world) and blood products. It is also of concern for persons receiving medical care in regions where there is prevalent substandard hygiene in the use of injection equipment, such as the reuse of needles in Third World countries. Health care workers such as nurses, laboratory workers, and doctors have also been infected, although this occurs more rarely. Since transmission of HIV by blood became known medical personnel are required to protect themselves from contact with blood by the use of universal precautions. People who give and receive tattoos, piercings, and scarification procedures can also be at risk of infection.
HIV has been found at low concentrations in the saliva, tears and urine of infected individuals, but there are no recorded cases of infection by these secretions and the potential risk of transmission is negligible.[50] It is not possible for mosquitoes to transmit HIV.[51]
Mother-to-child
The transmission of the virus from the mother to the child can occur in utero (during pregnancy), intrapartum (at childbirth), or via breast feeding. In the absence of treatment, the transmission rate up to birth between the mother and child is around 25%.[32] However, where combination antiretroviral drug treatment and Cesarian section are available, this risk can be reduced to as low as one percent.[32] Postnatal mother-to-child transmission may be largely prevented by complete avoidance of breast feeding; however, this has significant associated morbidity. Exclusive breast feeding and the provision of extended antiretroviral prophylaxis to the infant are also efficacious in avoiding transmission.[52]
Multiple infection
Unlike some other viruses, infection with HIV does not provide immunity against additional infections, particularly in the case of more genetically distant viruses. Both inter- and intra-clade multiple infections have been reported,[53] and even associated with more rapid disease progression.[54] Multiple infections are divided into two categories depending on the timing of the acquisition of the second strain. Coinfection refers to two strains that appear to have been acquired at the same time (or too close to distinguish). Reinfection (or superinfection) is infection with a second strain at a measurable time after the first. Both forms of dual infection have been reported for HIV in both acute and chronic infection around the world.[55][56][57][58]
Structure and genome

HIV is different in structure from other retroviruses. It is roughly spherical[59] with a diameter of about 120 nm, around 60 times smaller than a red blood cell, yet large for a virus.[60] It is composed of two copies of positive single-stranded RNA that codes for the virus's nine genes enclosed by a conical capsid composed of 2,000 copies of the viral protein p24.[61] The single-stranded RNA is tightly bound to nucleocapsid proteins, p7 and enzymes needed for the development of the virion such as reverse transcriptase, proteases, ribonuclease and integrase. A matrix composed of the viral protein p17 surrounds the capsid ensuring the integrity of the virion particle.[61]
This is, in turn, surrounded by the viral envelope that is composed of two layers of fatty molecules called phospholipids taken from the membrane of a human cell when a newly formed virus particle buds from the cell. Embedded in the viral envelope are proteins from the host cell and about 70 copies of a complex HIV protein that protrudes through the surface of the virus particle.[61] This protein, known as Env, consists of a cap made of three molecules called glycoprotein (gp) 120, and a stem consisting of three gp41 molecules that anchor the structure into the viral envelope.[62] This glycoprotein complex enables the virus to attach to and fuse with target cells to initiate the infectious cycle.[62] Both these surface proteins, especially gp120, have been considered as targets of future treatments or vaccines against HIV.[63]
The RNA genome consists of at least seven structural landmarks (LTR, TAR, RRE, PE, SLIP, CRS, and INS) and nine genes (gag, pol, and env, tat, rev, nef, vif, vpr, vpu, and sometimes a tenth tev, which is a fusion of tat env and rev) encoding 19 proteins. Three of these genes, gag, pol, and env, contain information needed to make the structural proteins for new virus particles.[61] For example, env codes for a protein called gp160 that is broken down by a viral enzyme to form gp120 and gp41. The six remaining genes, tat, rev, nef, vif, vpr, and vpu (or vpx in the case of HIV-2), are regulatory genes for proteins that control the ability of HIV to infect cells, produce new copies of virus (replicate), or cause disease.[61]
The two Tat proteins (p16 and p14) are transcriptional transactivators for the LTR promoter acting by binding the TAR RNA element. The TAR may also be processed into microRNAs that regulate the apoptosis genes ERCC1 and IER3.[64][65] The Rev protein (p19) is involved in shuttling RNAs from the nucleus and the cytoplasm by binding to the RRE RNA element. The Vif protein (p23) prevents the action of APOBEC3G (a cell protein that deaminates DNA:RNA hybrids and/or interferes with the Pol protein). The Vpr protein (p14) arrests cell division at G2/M. The Nef protein (p27) down-regulates CD4 (the major viral receptor), as well as the MHC class I and class II molecules.[66][67][68]
Nef also interacts with SH3 domains. The Vpu protein (p16) influences the release of new virus particles from infected cells.[61] The ends of each strand of HIV RNA contain an RNA sequence called the long terminal repeat (LTR). Regions in the LTR act as switches to control production of new viruses and can be triggered by proteins from either HIV or the host cell. The Psi element is involved in viral genome packaging and recognized by Gag and Rev proteins. The SLIP element (TTTTTT) is involved in the frameshift in the Gag-Pol reading frame required to make functional Pol.[61]
Tropism
The term viral tropism refers to which cell types HIV infects. HIV can infect a variety of immune cells such as CD4+ T cells, macrophages, and microglial cells. HIV-1 entry to macrophages and CD4+ T cells is mediated through interaction of the virion envelope glycoproteins (gp120) with the CD4 molecule on the target cells and also with chemokine coreceptors.[62]
Macrophage (M-tropic) strains of HIV-1, or non-syncitia-inducing strains (NSI) use the β-chemokine receptor CCR5 for entry and are thus able to replicate in macrophages and CD4+ T cells.[69] This CCR5 coreceptor is used by almost all primary HIV-1 isolates regardless of viral genetic subtype. Indeed, macrophages play a key role in several critical aspects of HIV infection. They appear to be the first cells infected by HIV and perhaps the source of HIV production when CD4+ cells become depleted in the patient. Macrophages and microglial cells are the cells infected by HIV in the central nervous system. In tonsils and adenoids of HIV-infected patients, macrophages fuse into multinucleated giant cells that produce huge amounts of virus.
T-tropic isolates, or syncitia-inducing (SI) strains replicate in primary CD4+ T cells as well as in macrophages and use the α-chemokine receptor, CXCR4, for entry.[69][70][71] Dual-tropic HIV-1 strains are thought to be transitional strains of HIV-1 and thus are able to use both CCR5 and CXCR4 as co-receptors for viral entry.
The α-chemokine SDF-1, a ligand for CXCR4, suppresses replication of T-tropic HIV-1 isolates. It does this by down-regulating the expression of CXCR4 on the surface of these cells. HIV that use only the CCR5 receptor are termed R5; those that only use CXCR4 are termed X4, and those that use both, X4R5. However, the use of coreceptor alone does not explain viral tropism, as not all R5 viruses are able to use CCR5 on macrophages for a productive infection[69] and HIV can also infect a subtype of myeloid dendritic cells,[72] which probably constitute a reservoir that maintains infection when CD4+ T cell numbers have declined to extremely low levels.
Some people are resistant to certain strains of HIV.[73] For example people with the CCR5-Δ32 mutation are resistant to infection with R5 virus as the mutation stops HIV from binding to this coreceptor, reducing its ability to infect target cells.
Sexual intercourse is the major mode of HIV transmission. Both X4 and R5 HIV are present in the seminal fluid, which is passed from a male to his sexual partner. The virions can then infect numerous cellular targets and disseminate into the whole organism. However, a selection process leads to a predominant transmission of the R5 virus through this pathway.[74][75][76] How this selective process works is still under investigation, but one model is that spermatozoa may selectively carry R5 HIV as they possess both CCR3 and CCR5 but not CXCR4 on their surface[77] and that genital epithelial cells preferentially sequester X4 virus.[78] In patients infected with subtype B HIV-1, there is often a co-receptor switch in late-stage disease and T-tropic variants appear that can infect a variety of T cells through CXCR4.[79] These variants then replicate more aggressively with heightened virulence that causes rapid T cell depletion, immune system collapse, and opportunistic infections that mark the advent of AIDS.[80] Thus, during the course of infection, viral adaptation to the use of CXCR4 instead of CCR5 may be a key step in the progression to AIDS. A number of studies with subtype B-infected individuals have determined that between 40 and 50% of AIDS patients can harbour viruses of the SI, and presumably the X4, phenotype.[81][82]
HIV-2 is much less pathogenic than HIV-1 and is restricted in its worldwide distribution. The adoption of "accessory genes" by HIV-2 and its more promiscuous pattern of coreceptor usage (including CD4-independence) may assist the virus in its adaptation to avoid innate restriction factors present in host cells. Adaptation to use normal cellular machinery to enable transmission and productive infection has also aided the establishment of HIV-2 replication in humans. A survival strategy for any infectious agent is not to kill its host but ultimately become a commensal organism. Having achieved a low pathogenicity, over time, variants more successful at transmission will be selected.[83]
Replication cycle

Entry to the cell
HIV enters macrophages and CD4+ T cells by the adsorption of glycoproteins on its surface to receptors on the target cell followed by fusion of the viral envelope with the cell membrane and the release of the HIV capsid into the cell.[84][85]
Entry to the cell begins through interaction of the trimeric envelope complex (gp160 spike) and both CD4 and a chemokine receptor (generally either CCR5 or CXCR4, but others are known to interact) on the cell surface.[84][85] gp120 binds to integrin α4β7 activating LFA-1 the central integrin involved in the establishment of virological synapses, which facilitate efficient cell-to-cell spreading of HIV-1.[86] The gp160 spike contains binding domains for both CD4 and chemokine receptors.[84][85]
The first step in fusion involves the high-affinity attachment of the CD4 binding domains of gp120 to CD4. Once gp120 is bound with the CD4 protein, the envelope complex undergoes a structural change, exposing the chemokine binding domains of gp120 and allowing them to interact with the target chemokine receptor.[84][85] This allows for a more stable two-pronged attachment, which allows the N-terminal fusion peptide gp41 to penetrate the cell membrane.[84][85] Repeat sequences in gp41, HR1 and HR2 then interact, causing the collapse of the extracellular portion of gp41 into a hairpin. This loop structure brings the virus and cell membranes close together, allowing fusion of the membranes and subsequent entry of the viral capsid.[84][85]
After HIV has bound to the target cell, the HIV RNA and various enzymes, including reverse transcriptase, integrase, ribonuclease, and protease, are injected into the cell.[84] During the microtubule based transport to the nucleus, the viral single strand RNA genome is transcribed into double strand DNA, which is then integrated into a host chromosome.
HIV can infect dendritic cells (DCs) by this CD4-CCR5 route, but another route using mannose-specific C-type lectin receptors such as DC-SIGN can also be used.[87] DCs are one of the first cells encountered by the virus during sexual transmission. They are currently thought to play an important role by transmitting HIV to T-cells when the virus is captured in the mucosa by DCs.[87] The presence of FEZ-1, which occurs naturally in neurons, is believed to prevent the infection of cells by HIV.[88]
Replication and transcription
Shortly after the viral capsid enters the cell, an enzyme called reverse transcriptase liberates the single-stranded (+)RNA genome from the attached viral proteins and copies it into a complementary DNA (cDNA) molecule.[89] The process of reverse transcription is extremely error-prone, and the resulting mutations may cause drug resistance or allow the virus to evade the body's immune system. The reverse transcriptase also has ribonuclease activity that degrades the viral RNA during the synthesis of cDNA, as well as DNA-dependent DNA polymerase activity that creates a sense DNA from the antisense cDNA.[90] Together, the cDNA and its complement form a double-stranded viral DNA that is then transported into the cell nucleus. The integration of the viral DNA into the host cell's genome is carried out by another viral enzyme called integrase.[89]
This integrated viral DNA may then lie dormant, in the latent stage of HIV infection.[89] To actively produce the virus, certain cellular transcription factors need to be present, the most important of which is NF-κB (NF kappa B), which is upregulated when T-cells become activated.[91] This means that those cells most likely to be killed by HIV are those currently fighting infection.
During viral replication, the integrated DNA provirus is transcribed into mRNA, which is then spliced into smaller pieces. These small pieces are exported from the nucleus into the cytoplasm, where they are translated into the regulatory proteins Tat (which encourages new virus production) and Rev. As the newly produced Rev protein accumulates in the nucleus, it binds to viral mRNAs and allows unspliced RNAs to leave the nucleus, where they are otherwise retained until spliced.[92] At this stage, the structural proteins Gag and Env are produced from the full-length mRNA. The full-length RNA is actually the virus genome; it binds to the Gag protein and is packaged into new virus particles.
HIV-1 and HIV-2 appear to package their RNA differently; HIV-1 will bind to any appropriate RNA, whereas HIV-2 will preferentially bind to the mRNA that was used to create the Gag protein itself. This may mean that HIV-1 is better able to mutate (HIV-1 infection progresses to AIDS faster than HIV-2 infection and is responsible for the majority of global infections).
Assembly and release
The final step of the viral cycle, assembly of new HIV-1 virons, begins at the plasma membrane of the host cell. The Env polyprotein (gp160) goes through the endoplasmic reticulum and is transported to the Golgi complex where it is cleaved by protease and processed into the two HIV envelope glycoproteins gp41 and gp120. These are transported to the plasma membrane of the host cell where gp41 anchors the gp120 to the membrane of the infected cell. The Gag (p55) and Gag-Pol (p160) polyproteins also associate with the inner surface of the plasma membrane along with the HIV genomic RNA as the forming virion begins to bud from the host cell. Maturation either occurs in the forming bud or in the immature virion after it buds from the host cell. During maturation, HIV proteases cleave the polyproteins into individual functional HIV proteins and enzymes. The various structural components then assemble to produce a mature HIV virion.[93] This cleavage step can be inhibited by protease inhibitors. The mature virus is then able to infect another cell.
Genetic variability

HIV differs from many viruses in that it has very high genetic variability. This diversity is a result of its fast replication cycle, with the generation of about 1010 virions every day, coupled with a high mutation rate of approximately 3 x 10−5 per nucleotide base per cycle of replication and recombinogenic properties of reverse transcriptase.[94][95][96]
This complex scenario leads to the generation of many variants of HIV in a single infected patient in the course of one day.[94] This variability is compounded when a single cell is simultaneously infected by two or more different strains of HIV. When simultaneous infection occurs, the genome of progeny virions may be composed of RNA strands from two different strains. This hybrid virion then infects a new cell where it undergoes replication. As this happens, the reverse transcriptase, by jumping back and forth between the two different RNA templates, will generate a newly synthesized retroviral DNA sequence that is a recombinant between the two parental genomes.[94] This recombination is most obvious when it occurs between subtypes.[94]
The closely related simian immunodeficiency virus (SIV) has evolved into many strains, classified by the natural host species. SIV strains of the African green monkey (SIVagm) and sooty mangabey (SIVsmm) are thought to have a long evolutionary history with their hosts. These hosts have adapted to the presence of the virus,[97] which is present at high levels in the host's blood but evokes only a mild immune response,[98] does not cause the development of simian AIDS,[99] and does not undergo the extensive mutation and recombination typical of HIV infection in humans.[100]
In contrast, when these strains infect species that have not adapted to SIV ("heterologous" hosts such as rhesus or cynomologus macaques), the animals develop AIDS and the virus generates genetic diversity similar to what is seen in human HIV infection.[101] Chimpanzee SIV (SIVcpz), the closest genetic relative of HIV-1, is associated with increased mortality and AIDS-like symptoms in its natural host.[102] Both SIVcpz and HIV-1 appear to have been transmitted relatively recently to chimpanzee and human populations, so their hosts have not yet adapted to the virus.[97] Both viruses have also lost a function of the Nef gene that is present in most SIVs; without this function, T cell depletion is more likely, leading to immunodeficiency.[102]
Three groups of HIV-1 have been identified on the basis of differences in the envelope (env) region: M, N, and O.[103] Group M is the most prevalent and is subdivided into eight subtypes (or clades), based on the whole genome, which are geographically distinct.[104] The most prevalent are subtypes B (found mainly in North America and Europe), A and D (found mainly in Africa), and C (found mainly in Africa and Asia); these subtypes form branches in the phylogenetic tree representing the lineage of the M group of HIV-1. Coinfection with distinct subtypes gives rise to circulating recombinant forms (CRFs). In 2000, the last year in which an analysis of global subtype prevalence was made, 47.2% of infections worldwide were of subtype C, 26.7% were of subtype A/CRF02_AG, 12.3% were of subtype B, 5.3% were of subtype D, 3.2% were of CRF_AE, and the remaining 5.3% were composed of other subtypes and CRFs.[105] Most HIV-1 research is focused on subtype B; few laboratories focus on the other subtypes.[106] The existence of a fourth group, "P", has been hypothesised based on a virus isolated in 2009.[107][108][109] The strain is apparently derived from gorilla SIV (SIVgor), first isolated from western lowland gorillas in 2006.[107]
The genetic sequence of HIV-2 is only partially homologous to HIV-1 and more closely resembles that of SIVsmm.
Diagnosis
Many HIV-positive people are unaware that they are infected with the virus.[110] For example, less than 1% of the sexually active urban population in Africa have been tested and this proportion is even lower in rural populations.[110] Furthermore, only 0.5% of pregnant women attending urban health facilities are counselled, tested or receive their test results.[110] Again, this proportion is even lower in rural health facilities.[110] Since donors may therefore be unaware of their infection, donor blood and blood products used in medicine and medical research are routinely screened for HIV.[111]
HIV-1 testing consists of initial screening with an enzyme-linked immunosorbent assay (ELISA) to detect antibodies to HIV-1. Specimens with a nonreactive result from the initial ELISA are considered HIV-negative unless new exposure to an infected partner or partner of unknown HIV status has occurred. Specimens with a reactive ELISA result are retested in duplicate.[112] If the result of either duplicate test is reactive, the specimen is reported as repeatedly reactive and undergoes confirmatory testing with a more specific supplemental test (e.g., Western blot or, less commonly, an immunofluorescence assay (IFA)). Only specimens that are repeatedly reactive by ELISA and positive by IFA or reactive by Western blot are considered HIV-positive and indicative of HIV infection. Specimens that are repeatedly ELISA-reactive occasionally provide an indeterminate Western blot result, which may be either an incomplete antibody response to HIV in an infected person, or nonspecific reactions in an uninfected person.[113]
Although IFA can be used to confirm infection in these ambiguous cases, this assay is not widely used. Generally, a second specimen should be collected more than a month later and retested for persons with indeterminate Western blot results. Although much less commonly available, nucleic acid testing (e.g., viral RNA or proviral DNA amplification method) can also help diagnosis in certain situations.[112] In addition, a few tested specimens might provide inconclusive results because of a low quantity specimen. In these situations, a second specimen is collected and tested for HIV infection.
Modern HIV testing is extremely accurate. The chance of a false-positive result in the two-step testing protocol is estimated to be 0.0004% to 0.0007% in the general U.S. population.[114][115][116][117]
Treatment
_300mg.jpg)
There is currently no publicly available vaccine or cure for HIV or AIDS.[118][119] However, a vaccine that is a combination of two previously unsuccessful vaccine candidates was reported in September 2009 to have resulted in a 30% reduction in infections in a trial conducted in Thailand.[120] Additionally, a course of antiretroviral treatment administered immediately after exposure, referred to as post-exposure prophylaxis, is believed to reduce the risk of infection if begun as quickly as possible.[121] In July 2010, a vaginal gel containing tenofovir, a reverse transcriptase inhibitor, was shown to reduce HIV infection rates by 39 percent in a trial conducted in South Africa.[122]
However, due to the incomplete protection provided by the vaccine and/or post-exposure prophylaxis, the avoidance of exposure to the virus is expected to remain the only reliable way to escape infection for some time yet. Current treatment for HIV infection consists of highly active antiretroviral therapy, or HAART.[123] This has been highly beneficial to many HIV-infected individuals since its introduction in 1996, when the protease inhibitor-based HAART initially became available.[124] Current HAART options are combinations (or "cocktails") consisting of at least three drugs belonging to at least two types, or "classes," of antiretroviral agents. Typically, these classes are two nucleoside analogue reverse transcriptase inhibitors (NARTIs or NRTIs) plus either a protease inhibitor or a non-nucleoside reverse transcriptase inhibitor (NNRTI).
New classes of drugs such as entry inhibitors provide treatment options for patients who are infected with viruses already resistant to common therapies, although they are not widely available and not typically accessible in resource-limited settings. Because AIDS progression in children is more rapid and less predictable than in adults, particularly in young infants, more aggressive treatment is recommended for children than adults.[125] In developed countries where HAART is available, doctors assess their patients thoroughly: measuring the viral load, how fast CD4 declines, and patient readiness. They then decide when to recommend starting treatment.[126]
HAART neither cures the patient nor does it uniformly remove all symptoms; high levels of HIV-1, often HAART resistant, return if treatment is stopped.[127][128] Moreover, it would take more than a lifetime for HIV infection to be cleared using HAART.[129] Despite this, many HIV-infected individuals have experienced remarkable improvements in their general health and quality of life, which has led to a large reduction in HIV-associated morbidity and mortality in the developed world.[124][130][131] One study suggests the average life expectancy of an HIV infected individual is 32 years from the time of infection if treatment is started when the CD4 count is 350/µL.[132]
In the absence of HAART, progression from HIV infection to AIDS has been observed to occur at a median of between nine to ten years and the median survival time after developing AIDS is only 9.2 months.[15] However, HAART sometimes achieves far less than optimal results, in some circumstances being effective in less than fifty percent of patients. This is due to a variety of reasons such as medication intolerance/side effects, prior ineffective antiretroviral therapy and infection with a drug-resistant strain of HIV. However, non-adherence and non-persistence with antiretroviral therapy is the major reason most individuals fail to benefit from HAART.[133]
The reasons for non-adherence and non-persistence with HAART are varied and overlapping. Major psychosocial issues, such as poor access to medical care, inadequate social supports, psychiatric disease and drug abuse contribute to non-adherence. The complexity of these HAART regimens, whether due to pill number, dosing frequency, meal restrictions or other issues along with side effects that create intentional non-adherence also contribute to this problem.[134][135][136] The side effects include lipodystrophy, dyslipidemia, insulin resistance, an increase in cardiovascular risks, and birth defects.[137][138]
The timing for starting HIV treatment is still debated. There is no question that treatment should be started before the patient's CD4 count falls below 200, and most national guidelines say to start treatment once the CD4 count falls below 350; but there is some evidence from cohort studies that treatment should be started before the CD4 count falls below 350.[130][139] In those countries where CD4 counts are not available, patients with WHO stage III or IV disease[140] should be offered treatment.
Anti-retroviral drugs are expensive, and the majority of the world's infected individuals do not have access to medications and treatments for HIV and AIDS.[141] Research to improve current treatments includes decreasing side effects of current drugs, further simplifying drug regimens to improve adherence, and determining the best sequence of regimens to manage drug resistance. Unfortunately, only a vaccine is thought to be able to halt the pandemic. This is because a vaccine would cost less, thus being affordable for developing countries, and would not require daily treatment.[141] However, after over 20 years of research, HIV-1 remains a difficult target for a vaccine.[141]
Treatments in development
Media reports in 2008 and a publication in the New England Journal of Medicine in 2009 described the anecdotal case of an HIV-positive patient of a Berlin doctor, Gero Hütter. The patient, who had both acute myelogenous leukemia (AML) and HIV infection, was said by some to be "functionally cured" of his HIV following a bone marrow transplant for AML. The bone marrow donor had been selected as homozygous for a CCR5-Δ32 mutation (which confers resistance to "almost all strains of HIV").[142][143] After 600 days without antiretroviral drug treatment, HIV levels in the patient's blood, bone marrow and bowel were below the limit of detection, although the authors note that the virus is likely present in other tissues. Researchers cautioned that it would be premature to consider this treatment a possible cure because of its anecdotal nature, the mortality risk associated with bone marrow transplants and other concerns.[144][145]
In 2010, a chemical called BanLec was reported to be a potent inhibitor of HIV replication.[146][147] Researchers at the University of Michigan determined that BanLec bound to the HIV-1 envelope protein gp120, which is high in sugar content, inhibiting viral entry into human cells.[146][147] The researchers suggest that such an inhibitor of HIV infection may find use as a topical treatment, such as a vaginal microbicide, and may be cheaper to produce than current antiviral topical treatments.[148] BanLec is not FDA approved for the treatment of HIV-1 infection and it should not be used to treat or prevent HIV-1 infection.
HIV latent reservoir
Despite the success of highly active antiretroviral therapy (HAART) in controlling HIV infection and reducing HIV-associated mortality, current drug regimens are unable to completely eradicate HIV infection. Many people on HAART achieve suppression of HIV to levels below the limit of detection of standard clinical assays for many years. However, upon withdrawal of HAART, HIV viral loads rebound quickly with a concomitant decline in CD4+ T-Cells, which, in most cases, absent a resumption of treatment, leads to AIDS.
To successfully reproduce itself, HIV must convert its RNA genome to DNA, which is then imported into the host cell's nucleus and inserted into the host genome through the action of HIV integrase. Because HIV's primary cellular target, CD4+ T-Cells, function as the memory cells of the immune system, integrated HIV can remain dormant for the duration of these cell's lifetime. Memory T-Cells may survive for many years and possibly for decades. The latent HIV reservoir can be measured by co-culturing CD4+ T-Cells from infected patients with CD4+ T-Cells from uninfected donors and measuring HIV protein or RNA.[149]
The failure of vaccine candidates to protect against HIV infection and progression to AIDS has led to a renewed focus on the biological mechanisms responsible for HIV latency. A limited period of therapy combining anti-retrovirals with drugs targeting the latent reservoir may one day allow for total eradication of HIV infection.[150]
Prognosis
Without treatment, the net median survival time after infection with HIV is estimated to be 9 to 11 years, depending on the HIV subtype,[151] and the median survival rate after diagnosis of AIDS in resource-limited settings where treatment is not available ranges between 6 and 19 months, depending on the study.[152] In areas where it is widely available, the development of HAART as effective therapy for HIV infection and AIDS reduced the death rate from this disease by 80%, and raised the life expectancy for a newly diagnosed HIV-infected person to 20–50 years.[153][154]
As new treatments continue to be developed and because HIV continues to evolve resistance to treatments, estimates of survival time are likely to continue to change. Without antiretroviral therapy, death normally occurs within a year after the individual progresses to AIDS.[15] Most patients die from opportunistic infections or malignancies associated with the progressive failure of the immune system.[11] The rate of clinical disease progression varies widely between individuals and has been shown to be affected by many factors such as host susceptibility and immune function[73][155][156] health care and co-infections,[11][15] as well as which particular strain of the virus is involved.[157][158][159]
Epidemiology



UNAIDS and the WHO estimate that AIDS has killed more than 25 million people since it was first recognized in 1981, making it one of the most destructive pandemics in recorded history. Despite recent improved access to antiretroviral treatment and care in many regions of the world, the AIDS pandemic claimed an estimated 2.8 million (between 2.4 and 3.3 million) lives in 2005 of which more than half a million (570,000) were children.[5]
In 2007, between 30.6 and 36.1 million people were believed to live with HIV, and it killed an estimated 2.1 million people that year, including 330,000 children; there were 2.5 million new infections.[151]
Sub-Saharan Africa remains by far the worst-affected region, with an estimated 21.6 to 27.4 million people currently living with HIV. Two million (1.5–3.0 million) of them are children younger than 15 years of age. More than 64% of all people living with HIV are in sub-Saharan Africa, as are more than three quarters of all women living with HIV. In 2005, there were 12.0 million (10.6–13.6 million) AIDS orphans living in sub-Saharan Africa 2005.[5]
South & South East Asia are second-worst affected with 15% of the total. AIDS accounts for the deaths of 500,000 children in this region. South Africa has the largest number of HIV patients in the world followed by Nigeria.[161] Other Sub-Saharan countries, such as the Sudan report lower prevalences of 1.6%, although the data is poorly documented.[162] India has an estimated 2.5 million infections (0.23% of population), making India the country with the third largest population of HIV patients. In the 35 African nations with the highest prevalence, average life expectancy is 48.3 years—6.5 years less than it would be without the disease.[163]
The latest evaluation report of the World Bank's Operations Evaluation Department assesses the development effectiveness of the World Bank's country-level HIV/AIDS assistance defined as policy dialogue, analytic work, and lending with the explicit objective of reducing the scope or impact of the AIDS epidemic.[164] This is the first comprehensive evaluation of the World Bank's HIV/AIDS support to countries, from the beginning of the epidemic through mid-2004. Because the Bank aims to assist in implementation of national government programmes, their experience provides important insights on how national AIDS programmes can be made more effective.
The development of HAART as effective therapy for HIV infection has substantially reduced the death rate from this disease in those areas where these drugs are widely available.[124] As the life expectancy of persons with HIV has increased in countries where HAART is widely used, the continuing spread of the disease has caused the number of persons living with HIV to increase substantially.
In Africa, the number of mother-to-child-transmission (MTCT) cases and the prevalence of AIDS is beginning to reverse decades of steady progress in child survival. Countries such as Uganda are attempting to curb the MTCT epidemic by offering VCT (voluntary counselling and testing), PMTCT (prevention of mother-to-child transmission) and ANC (ante-natal care) services, which include the distribution of antiretroviral therapy.
History
Origins
- See History of known cases and spread for early cases of HIV / AIDS
HIV is thought to have originated in non-human primates in sub-Saharan Africa and was transferred to humans late in the 19th or early in the 20th century.[165] The first paper recognizing a pattern of opportunistic infections characteristic of AIDS was published in 1981.[166]
Both HIV-1 and HIV-2 are believed to have originated in West-Central Africa and to have jumped species (a process known as zoonosis) from non-human primates to humans. HIV-1 appears to have originated in southern Cameroon through the evolution of SIV(cpz), a simian immunodeficiency virus (SIV) that infects wild chimpanzees (Pan troglodytes troglodytes).[167][168] The closest relative of HIV-2 is SIV(agm), a virus of the sooty mangabey (Cercocebus atys), an Old World monkey of Guinea-Bissau, Gabon, and Cameroon.[21] New World monkeys such as the owl monkey are resistant to HIV-1 infection, possibly because of a genomic fusion of two viral resistance genes.[169]
Discovery
AIDS was first clinically observed between late 1980 and early 1981.[170] Injection drug users and gay men with no known cause of impaired immunity showed symptoms of Pneumocystis carinii pneumonia (PCP), a rare opportunistic infection that was known to present itself in people with very compromised immune systems.[171][172][173] Soon thereafter, additional gay men developed a previously-rare skin cancer called Kaposi’s sarcoma (KS).[174][175] Many more cases of PCP and KS quickly emerged, alerting U.S. Centers for Disease Control and Prevention (CDC). A CDC task force was formed to monitor the outbreak. After recognizing a pattern of anomalous symptoms presenting themselves in patients, the task force named the condition acquired immune deficiency syndrome (AIDS).[176]
In 1983, two separate research groups led by Robert Gallo and Luc Montagnier independently declared that a novel retrovirus may have been infecting AIDS patients, and published their findings in the same issue of the journal Science.[177][178] Gallo claimed that a virus his group had isolated from an AIDS patient was strikingly similar in shape to other human T-lymphotropic viruses (HTLVs) his group had been the first to isolate. Gallo's group called their newly isolated virus HTLV-III. At the same time, Montagnier's group isolated a virus from a patient presenting lymphadenopathy (swelling of the lymph nodes) of the neck and physical weakness, two classic symptoms of AIDS. Contradicting the report from Gallo's group, Montagnier and his colleagues showed that core proteins of this virus were immunologically different from those of HTLV-I. Montagnier's group named their isolated virus lymphadenopathy-associated virus (LAV).[176] HIV was chosen as a compromise between the two claims (LAV and HTLV-III).
Whether Gallo or Montagnier deserve more credit for the discovery of the virus that causes AIDS has been a matter of considerable controversy. Together with his colleague Françoise Barré-Sinoussi, Montagnier was awarded one half of the 2008 Nobel Prize in Physiology or Medicine for his "discovery of human immunodeficiency virus".[179] Harald zur Hausen also shared the Prize for his discovery that human papilloma virus leads to cervical cancer, but Gallo was left out.[180] Gallo said that it was "a disappointment" that he was not named a co-recipient.[181] Montagnier said he was "surprised" Gallo was not recognized by the Nobel Committee: "It was important to prove that HIV was the cause of AIDS, and Gallo had a very important role in that. I'm very sorry for Robert Gallo."[180]
AIDS denialism
Some individuals, including some scientists who are not recognized experts on HIV,[182] question the connection between HIV and AIDS,[183] the existence of HIV itself, or the validity of HIV testing and treatment methods.[182][184] These claims, known as AIDS denialism, have been examined and rejected by most of the worldwide scientific community,[185] although they have had a political impact, particularly in South Africa, where the government's official promotion of AIDS denialism was responsible for its ineffective response to that country's AIDS epidemic.[186][187][188]
See also
- HIV blood screening
- HIV disease-related drug reaction
- HIV-associated pruritus
- Photosensitivity with HIV infection
- Discovery and development of CCR5 receptor antagonists
References
- ↑ Weiss RA (May 1993). "How does HIV cause AIDS?". Science 260 (5112): 1273–9. doi:10.1126/science.8493571. PMID 8493571.
- ↑ Douek DC, Roederer M, Koup RA (2009). "Emerging concepts in the immunopathogenesis of AIDS". Annu. Rev. Med. 60: 471–84. doi:10.1146/annurev.med.60.041807.123549. PMID 18947296.
- ↑ 3.0 3.1 "CDC - HIV/AIDS - Resources - HIV Prevention in the United States at a Critical Crossroads". Cdc.gov. http://www.cdc.gov/hiv/resources/reports/hiv_prev_us.htm. Retrieved 2010-07-28.
- ↑ 4.0 4.1 "HIV and AIDS among Gay and Bisexual Men" (PDF). http://www.cdc.gov/nchhstp/newsroom/docs/FastFacts-MSM-FINAL508COMP.pdf. Retrieved 2010-07-28.
- ↑ 5.0 5.1 5.2 5.3 Joint United Nations Programme on HIV/AIDS (2006). "Overview of the global AIDS epidemic" (PDF). 2006 Report on the global AIDS epidemic. ISBN 9291734799. http://data.unaids.org/pub/GlobalReport/2006/2006_GR_CH02_en.pdf. Retrieved 2006-06-08.
- ↑ Greener, R. (2002). "AIDS and macroeconomic impact". In S, Forsyth (ed.). State of The Art: AIDS and Economics. IAEN. pp. 49–55. http://db.jhuccp.org/ics-wpd/exec/icswppro.dll?BU=http://db.jhuccp.org/ics-wpd/exec/icswppro.dll&QF0=DocNo&QI0=285428&TN=Popline&AC=QBE_QUERY&MR=30%25DL=1&&RL=1&&RF=LongRecordDisplay&DF=LongRecordDisplay.
- ↑ Joint United Nations Programme on HIV/AIDS. "AIDS epidemic update, 2005" (PDF). http://www.unaids.org/epi/2005/doc/EPIupdate2005_pdf_en/epi-update2005_en.pdf. Retrieved 2006-02-28.
- ↑ Palella, F. J. Jr, Delaney, K. M., Moorman, A. C., Loveless, M. O., Fuhrer, J., Satten, G. A., Aschman and D. J., Holmberg, S. D. (1998). "Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. HIV Outpatient Study Investigators". N. Engl. J. Med 338 (13): 853–860. doi:10.1056/NEJM199803263381301. PMID 9516219.
- ↑ PMID 20598938 (PubMed)
Citation will be completed automatically in a few minutes. Jump the queue or expand by hand - ↑ PMID 20628133 (PubMed)
Citation will be completed automatically in a few minutes. Jump the queue or expand by hand - ↑ 11.0 11.1 11.2 Lawn SD (2004). "AIDS in Africa: the impact of coinfections on the pathogenesis of HIV-1 infection". J. Infect. Dis. 48 (1): 1–12. doi:10.1016/j.jinf.2003.09.001. PMID 14667787.
- ↑ Buchbinder SP, Katz MH, Hessol NA, O'Malley PM, Holmberg SD. (1994). "Long-term HIV-1 infection without immunologic progression". AIDS 8 (8): 1123–8. doi:10.1097/00002030-199408000-00014. PMID 7986410.
- ↑ "Time from HIV-1 seroconversion to AIDS and death before widespread use of highly active antiretroviral therapy: a collaborative re-analysis. Collaborative Group on AIDS Incubation and HIV Survival including the CASCADE EU Concerted Action. Concerted Action on SeroConversion to AIDS and Death in Europe". Lancet 355 (9210): 1131–7. April 2000. doi:10.1016/S0140-6736(00)02061-4. PMID 10791375.
- ↑ Schneider MF, Gange SJ, Williams CM, Anastos K, Greenblatt RM, Kingsley L, Detels R, Munoz A (2005). "Patterns of the hazard of death after AIDS through the evolution of antiretroviral therapy: 1984–2004". AIDS 19 (17): 2009–18. doi:10.1097/01.aids.0000189864.90053.22. PMID 16260908.
- ↑ 15.0 15.1 15.2 15.3 Morgan D, Mahe C, Mayanja B, Okongo JM, Lubega R, Whitworth JA (2002). "HIV-1 infection in rural Africa: is there a difference in median time to AIDS and survival compared with that in industrialized countries?". AIDS 16 (4): 597–632. doi:10.1097/00002030-200203080-00011. PMID 11873003.
- ↑ International Committee on Taxonomy of Viruses. "61.0.6. Lentivirus". National Institutes of Health. http://www.ncbi.nlm.nih.gov/ICTVdb/ICTVdB/61060000.htm. Retrieved 2006-02-28.
- ↑ International Committee on Taxonomy of Viruses. "61. Retroviridae". National Institutes of Health. http://www.ncbi.nlm.nih.gov/ICTVdb/ICTVdB/61000000.htm. Retrieved 2006-02-28.
- ↑ Lévy, J. A. (1993). "HIV pathogenesis and long-term survival". AIDS 7 (11): 1401–10. doi:10.1097/00002030-199311000-00001. PMID 8280406.
- ↑ Smith, Johanna A.; Daniel, René (Division of Infectious Diseases, Center for Human Virology, Thomas Jefferson University, Philadelphia) (2006). "Following the path of the virus: the exploitation of host DNA repair mechanisms by retroviruses". ACS Chem Biol 1 (4): 217–26. doi:10.1021/cb600131q. PMID 17163676.
- ↑ Gilbert, PB et al; McKeague, IW; Eisen, G; Mullins, C; Guéye-Ndiaye, A; Mboup, S; Kanki, PJ (28 February 2003). "Comparison of HIV-1 and HIV-2 infectivity from a prospective cohort study in Senegal". Statistics in Medicine 22 (4): 573–593. doi:10.1002/sim.1342. PMID 12590415.
- ↑ 21.0 21.1 Reeves, J. D. and Doms, R. W (2002). "Human Immunodeficiency Virus Type 2". J. Gen. Virol. 83 (Pt 6): 1253–65. PMID 12029140.
- ↑ Grabar, S., Selinger-Leneman, H., Abgrall, S., Pialoux, G., Weiss, L., Costagliola, D. (2009). "Prevalence and comparative characteristics of long-term nonprogressors and HIV controller patients in the French Hospital Database on HIV". AIDS 23 (9): 1163–1169. doi:10.1097/QAD.0b013e32832b44c8. PMID 19444075.
- ↑ Piatak, M., Jr, Saag, M. S., Yang, L. C., Clark, S. J., Kappes, J. C., Luk, K. C., Hahn, B. H., Shaw, G. M. and Lifson, J.D. (1993). "High levels of HIV-1 in plasma during all stages of infection determined by competitive PCR". Science 259 (5102): 1749–1754. doi:10.1126/science.8096089. PMID 8096089.
- ↑ Pantaleo G, Demarest JF, Schacker T, Vaccarezza M, Cohen OJ, Daucher M, Graziosi C, Schnittman SS, Quinn TC, Shaw GM, Perrin L, Tambussi G, Lazzarin A, Sekaly RP, Soudeyns H, Corey L, Fauci AS. (1997). "The qualitative nature of the primary immune response to HIV infection is a prognosticator of disease progression independent of the initial level of plasma viremia". Proc Natl Acad Sci U S A. 94 (1): 254–258. doi:10.1073/pnas.94.1.254. PMID 8990195.
- ↑ Kahn, J. O. and Walker, B. D. (1998). "Acute Human Immunodeficiency Virus type 1 infection". N. Engl. J. Med. 331 (1): 33–39. doi:10.1056/NEJM199807023390107. PMID 9647878.
- ↑ Daar ES, Little S, Pitt J, et al. (2001). "Diagnosis of primary HIV-1 infection. Los Angeles County Primary HIV Infection Recruitment Network". Ann. Intern. Med. 134 (1): 25–9. PMID 11187417.
- ↑ Burton GF, Keele BF, Estes JD, Thacker TC, Gartner S. (2002). "Follicular dendritic cell contributions to HIV pathogenesis". Semin Immunol. 14 (4): 275–284. doi:10.1016/S1044-5323(02)00060-X. PMID 12163303.
- ↑ Clapham PR, McKnight A. (2001). "HIV-1 receptors and cell tropism". Br Med Bull. 58 (4): 43–59. doi:10.1093/bmb/58.1.43. PMID 11714623.
- ↑ Smith DK, Grohskopf LA, Black RJ, et al. (January 2005). "Antiretroviral postexposure prophylaxis after sexual, injection-drug use, or other nonoccupational exposure to HIV in the United States: recommendations from the U.S. Department of Health and Human Services". MMWR Recomm Rep 54 (RR-2): 1–20. PMID 15660015. http://www.cdc.gov/mmwr/preview/mmwrhtml/rr5402a1.htm#tab1. Retrieved 2009-03-31.
- ↑ 30.0 30.1 30.2 30.3 Jin F et al. (March 2010). "Per-contact probability of HIV transmission in homosexual men in Sydney in the era of HAART". AIDS 24 (6): 907–913. doi:10.1097/QAD.0b013e3283372d90. PMID 20139750.
- ↑ Donegan E, Stuart M, Niland JC, et al. (1990). "Infection with human immunodeficiency virus type 1 (HIV-1) among recipients of antibody-positive blood donations". Ann. Intern. Med. 113 (10): 733–739. PMID 2240875.
- ↑ 32.0 32.1 32.2 Coovadia H (2004). "Antiretroviral agents—how best to protect infants from HIV and save their mothers from AIDS". N. Engl. J. Med. 351 (3): 289–292. doi:10.1056/NEJMe048128. PMID 15247337.
- ↑ Kaplan EH, Heimer R (1995). "HIV incidence among New Haven needle exchange participants: updated estimates from syringe tracking and testing data". J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 10 (2): 175–176. PMID 7552482.
- ↑ Bell DM (1997). "Occupational risk of human immunodeficiency virus infection in healthcare workers: an overview.". Am. J. Med. 102 (5B): 9–15. doi:10.1016/S0002-9343(97)89441-7. PMID 9845490.
- ↑ 35.0 35.1 35.2 35.3 35.4 35.5 Boily MC, Baggaley RF, Wang L, Masse B, White RG, Hayes RJ, Alary M (February 2009). "Heterosexual risk of HIV-1 infection per sexual act: systematic review and meta-analysis of observational studies". The Lancet Infectious Diseases 9 (2): 118–129. doi:10.1016/S1473-3099(09)70021-0. PMID 19179227.
- ↑ 36.0 36.1 36.2 36.3 European Study Group on Heterosexual Transmission of HIV (1992). "Comparison of female to male and male to female transmission of HIV in 563 stable couples". BMJ. 304 (6830): 809–813. doi:10.1136/bmj.304.6830.809. PMID 1392708.
- ↑ 37.0 37.1 37.2 37.3 37.4 37.5 Varghese B, Maher JE, Peterman TA, Branson BM,Steketee RW (2002). "Reducing the risk of sexual HIV transmission: quantifying the per-act risk for HIV on the basis of choice of partner, sex act, and condom use". Sex. Transm. Dis. 29 (1): 38–43. PMID 11773877.
- ↑ Leynaert B, Downs AM, de Vincenzi I (1998). "Heterosexual transmission of human immunodeficiency virus: variability of infectivity throughout the course of infection. European Study Group on Heterosexual Transmission of HIV". Am. J. Epidemiol. 148 (1): 88–96. PMID 9663408.
- ↑ 39.0 39.1 Millett GA, Flores SA, Marks G, Reed JB, Herbst JH (October 2009). "Circumcision status and risk of HIV and sexually transmitted infections among men who have sex with men: a meta-analysis". The Journal of American Medical Association 300 (14): 1674–1684. doi:10.1001/jama.300.14.1674. PMID 18840841. http://jama.ama-assn.org/cgi/content/short/300/14/1674. Retrieved 2010-04-11.
- ↑ Correction about the values although "the pattern of nonsignificant findings remains consistent with the originally published article"[1]
- ↑ "Public Health Agency of Canada". Phac-aspc.gc.ca. 2004-12-01. http://www.phac-aspc.gc.ca/publicat/epiu-aepi/epi_update_may_04/13-eng.php. Retrieved 2010-07-28.
- ↑ National Institute of Allergy and Infectious Diseases; National Institutes of Health, Department of Health and Human Services (2001-07-20). "Workshop Summary: Scientific Evidence on Condom Effectiveness for Sexually Transmitted Disease (STD) Prevention" (PDF). Hyatt Dulles Airport, Herndon, Virginia. pp. 13–15. http://www3.niaid.nih.gov/about/organization/dmid/PDF/condomReport.pdf. Retrieved 2009-01-08.
- ↑ "Should spermicides be used with condoms?". Condoms and Sexually Transmitted Diseases, Brochure. United States Food and Drug Administration. 2009-04-30. http://www.fda.gov/ForConsumers/byAudience/ForPatientAdvocates/HIVandAIDSActivities/ucm126372.htm. Retrieved 2009-07-23.
- ↑ Global Campaign for Microbicides : Rectal Use of N-9 checked 2009-07-22
- ↑ Nonoxynol-9 Spermicide on HIV Risk List checked 2009-07-22
- ↑ Williams BG, Lloyd-Smith JO, Gouws E, Hankins C, Getz WM, Hargrove J, de Zoysa I, Dye C, Auvert B. (2006). "The Potential Impact of Male Circumcision on HIV in Sub-Saharan Africa.". PLoS Med 3 (7): e262. doi:10.1371/journal.pmed.0030262. PMID 16822094.
- ↑ Bailey RC, Moses S, Parker CB, et al. (2007). "Male circumcision for HIV prevention in young men in Kisumu, Kenya: a randomised controlled trial". Lancet 369 (9562): 643–56. doi:10.1016/S0140-6736(07)60312-2. PMID 17321310.
- ↑ Gray RH et al. (February 24, 2007). "Male circumcision for HIV prevention in men in Rakai, Uganda: a randomised trial". Lancet 369 (9562): 657–66. doi:10.1016/S0140-6736(07)60313-4. PMID 17321311.
- ↑ WHO (2007). "WHO and UNAIDS announce recommendations from expert consultation on male circumcision for HIV prevention". WHO.int. http://www.who.int/hiv/mediacentre/news68/en/index.html. Retrieved 2007-07-13.
- ↑ Lifson AR (1988). "Do alternate modes for transmission of human immunodeficiency virus exist? A review". JAMA 259 (9): 1353–6. doi:10.1001/jama.259.9.1353. PMID 2963151.
- ↑ "Why Mosquitoes Cannot Transmit AIDS [HIV virus]". Rci.rutgers.edu. http://www.rci.rutgers.edu/%7Einsects/aids.htm. Retrieved 2010-07-28.
- ↑ Cochrane Systematic Review on interventions for prevention of late postnatal mother-to-child transmission of HIV http://www.cochrane.org/reviews/en/ab006734.html
- ↑ Smith D, Richman D, Little S (2005). "HIV Superinfection". Journal of Infectious Diseases 192 (3): 438–44. doi:10.1086/431682. PMID 15995957.
- ↑ Gottlieb, et al.; Nickle, DC; Jensen, MA; Wong, KG; Grobler, J; Li, F; Liu, SL; Rademeyer, C et al. (2004). "Dual HIV-1 infection associated with rapid disease progression". Lancet 363 (9049): 619–22. doi:10.1016/S0140-6736(04)15596-7. PMID 14987889.
- ↑ Smith et al.; Wong, JK; Hightower, GK; Ignacio, CC; Koelsch, KK; Daar, ES; Richman, DD; Little, SJ (2004). "Incidence of HIV superinfection following primary infection". JAMA 292 (10): 1177–8. doi:10.1001/jama.292.10.1177. PMID 15353529.
- ↑ Chohan B, Lavreys L, Rainwater SM, Overbaugh J (August 2005). "Evidence for frequent reinfection with human immunodeficiency virus type 1 of a different subtype". J. Virol. 79 (16): 10701–8. doi:10.1128/JVI.79.16.10701-10708.2005. PMID 16051862.
- ↑ Piantadosi A, Chohan B, Chohan V, McClelland RS, Overbaugh J (November 2007). "Chronic HIV-1 infection frequently fails to protect against superinfection". PLoS Pathog. 3 (11): e177. doi:10.1371/journal.ppat.0030177. PMID 18020705.
- ↑ Hu DJ, Subbarao S, Vanichseni S, et al. (February 2005). "Frequency of HIV-1 dual subtype infections, including intersubtype superinfections, among injection drug users in Bangkok, Thailand". AIDS 19 (3): 303–8. PMID 15718841. http://meta.wkhealth.com/pt/pt-core/template-journal/lwwgateway/media/landingpage.htm?an=00002030-200502180-00009. Retrieved 2009-03-31.
- ↑ McGovern SL, Caselli E, Grigorieff N, Shoichet BK (2002). "A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening". J Med Chem 45 (8): 1712–22. doi:10.1021/jm010533y. PMID 11931626.
- ↑ Compared with overview in: Fisher, Bruce; Harvey, Richard P.; Champe, Pamela C. (2007). Lippincott's Illustrated Reviews: Microbiology (Lippincott's Illustrated Reviews Series). Hagerstown, MD: Lippincott Williams & Wilkins. ISBN 0-7817-8215-5. Page 3
- ↑ 61.0 61.1 61.2 61.3 61.4 61.5 61.6 Various (2008) (PDF). HIV Sequence Compendium 2008 Introduction. http://www.hiv.lanl.gov/content/sequence/HIV/COMPENDIUM/2008/frontmatter.pdf. Retrieved 2009-03-31.
- ↑ 62.0 62.1 62.2 Chan, DC., Fass, D., Berger, JM., Kim, PS. (1997). "Core Structure of gp41 from the HIV Envelope Glycoprotein" (PDF). Cell 89 (2): 263–73. doi:10.1016/S0092-8674(00)80205-6. PMID 9108481. http://www.its.caltech.edu/~chanlab/PDFs/Chan_Cell_1997.pdf. Retrieved 2009-03-31.
- ↑ National Institute of Health (June 17, 1998). "Crystal Structure of Key HIV Protein Reveals New Prevention, Treatment Targets". http://www3.niaid.nih.gov/news/newsreleases/1998/hivprotein.htm. Retrieved 2006-09-14.
- ↑ Ouellet DL, Plante I, Landry P, et al. (April 2008). "Identification of functional microRNAs released through asymmetrical processing of HIV-1 TAR element". Nucleic Acids Res. 36 (7): 2353–65. doi:10.1093/nar/gkn076. PMID 18299284. PMC 2367715. http://nar.oxfordjournals.org/cgi/content/full/36/7/2353.
- ↑ Klase Z, Winograd R, Davis J, et al. (2009). "HIV-1 TAR miRNA protects against apoptosis by altering cellular gene expression". Retrovirology 6: 18. doi:10.1186/1742-4690-6-18. PMID 19220914. PMC 2654423. http://www.retrovirology.com/content/6/1/18.
- ↑ Garcia JV, Miller AD (April 1991). "Serine phosphorylation-independent downregulation of cell-surface CD4 by nef". Nature 350 (6318): 508–11. doi:10.1038/350508a0. PMID 2014052.
- ↑ Schwartz O, Maréchal V, Le Gall S, Lemonnier F, Heard JM (March 1996). "Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein". Nat. Med. 2 (3): 338–42. doi:10.1038/nm0396-338. PMID 8612235.
- ↑ Stumptner-Cuvelette P, Morchoisne S, Dugast M, et al. (October 2001). "HIV-1 Nef impairs MHC class II antigen presentation and surface expression". Proc. Natl. Acad. Sci. U.S.A. 98 (21): 12144–9. doi:10.1073/pnas.221256498. PMID 11593029.
- ↑ 69.0 69.1 69.2 Coakley, E., Petropoulos, C. J. and Whitcomb, J. M. (2005). "Assessing ch vbgemokine co-receptor usage in HIV". Curr. Opin. Infect. Dis. 18 (1): 9–15. doi:10.1097/00001432-200502000-00003. PMID 15647694.
- ↑ Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, Di Marzio P, Marmon S, Sutton RE, Hill CM, Davis CB, Peiper SC, Schall TJ, Littman DR, Landau NR. (1996). "Identification of a major co-receptor for primary isolates of HIV-1". Nature 381 (6584): 661–6. doi:10.1038/381661a0. PMID 8649511.
- ↑ Feng Y, Broder CC, Kennedy PE, Berger EA. (1996). "HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor". Science 272 (5263): 872–7. doi:10.1126/science.272.5263.872. PMID 8629022.
- ↑ Knight, S. C., Macatonia, S. E. and Patterson, S. (1990). "HIV I infection of dendritic cells". Int. Rev. Immunol. 6 (2-3): 163–75. doi:10.3109/08830189009056627. PMID 2152500.
- ↑ 73.0 73.1 Tang, J. and Kaslow, R. A. (2003). "The impact of host genetics on HIV infection and disease progression in the era of highly active antiretroviral therapy". AIDS 17 (Suppl 4): S51–S60. doi:10.1097/00002030-200317004-00006. PMID 15080180.
- ↑ Zhu T, Mo H, Wang N, Nam DS, Cao Y, Koup RA, Ho DD. (1993). "Genotypic and phenotypic characterization of HIV-1 patients with primary infection". Science 261 (5125): 1179–81. doi:10.1126/science.8356453. PMID 8356453.
- ↑ van’t Wout AB, Kootstra NA, Mulder-Kampinga GA, Albrecht-van Lent N, Scherpbier HJ, Veenstra J, Boer K, Coutinho RA, Miedema F, Schuitemaker H. (1994). "Macrophage-tropic variants initiate human immunodeficiency virus type 1 infection after sexual, parenteral, and vertical transmission". J Clin Invest 94 (5): 2060–7. doi:10.1172/JCI117560. PMID 7962552.
- ↑ Zhu T, Wang N, Carr A, Nam DS, Moor-Jankowski R, Cooper DA, Ho DD. (1996). "Genetic characterization of human immunodeficiency virus type 1 in blood and genital secretions: evidence for viral compartmentalization and selection during sexual transmission". J Virol 70 (5): 3098–107. PMID 8627789.
- ↑ Muciaccia B, Padula F, Vicini E, Gandini L, Lenzi A, Stefanini M. (2005). "Beta-chemokine receptors 5 and 3 are expressed on the head region of human spermatozoon". FASEB J 19 (14): 2048–50. doi:10.1096/fj.05-3962fje. PMID 16174786.
- ↑ Berlier W, Bourlet T, Lawrence P, Hamzeh H, Lambert C, Genin C, Verrier B, Dieu-Nosjean MC, Pozzetto B, Delezay O. (2005). "Selective sequestration of X4 isolates by human genital epithelial cells: Implication for virus tropism selection process during sexual transmission of HIV". J Med Virol. 77 (4): 465–74. doi:10.1002/jmv.20478. PMID 16254974.
- ↑ Clevestig P, Maljkovic I, Casper C, Carlenor E, Lindgren S, Naver L, Bohlin AB, Fenyo EM, Leitner T, Ehrnst A. (2005). "The X4 phenotype of HIV type 1 evolves from R5 in two children of mothers, carrying X4, and is not linked to transmission". AIDS Res Hum Retroviruses 5 (21): 371–8. doi:10.1089/aid.2005.21.371. PMID 15929699.
- ↑ Moore JP. (1997). "Coreceptors: implications for HIV pathogenesis and therapy". Science 276 (5309): 51–2. doi:10.1126/science.276.5309.51. PMID 9122710.
- ↑ Karlsson A, Parsmyr K, Aperia K, Sandstrom E, Fenyo EM, Albert J. (1994). "MT-2 cell tropism of human immunodeficiency virus type 1 isolates as a marker for response to treatment and development of drug resistance". J Infect Dis. 170 (6): 1367–75. PMID 7995974.
- ↑ Koot M, van 't Wout AB, Kootstra NA, de Goede RE, Tersmette M, Schuitemaker H. (1996). "Relation between changes in cellular load, evolution of viral phenotype, and the clonal composition of virus populations in the course of human immunodeficiency virus type 1 infection". J Infect Dis. 173 (2): 349–54. PMID 8568295.
- ↑ Cheney, K and McKnight, A (2010). "HIV-2 Tropism and Disease". Lentiviruses and Macrophages: Molecular and Cellular Interactions. Caister Academic Press. ISBN 978-1-904455-60-8.
- ↑ 84.0 84.1 84.2 84.3 84.4 84.5 84.6 Chan D, Kim P (1998). "HIV entry and its inhibition". Cell 93 (5): 681–4. doi:10.1016/S0092-8674(00)81430-0. PMID 9630213.
- ↑ 85.0 85.1 85.2 85.3 85.4 85.5 Wyatt R, Sodroski J (1998). "The HIV-1 envelope glycoproteins: fusogens, antigens, and immunogens". Science 280 (5371): 1884–8. doi:10.1126/science.280.5371.1884. PMID 9632381.
- ↑ Arthos J, Cicala C, Martinelli E, Macleod K, Van Ryk D, Wei D, Xiao Z, Veenstra TD, Conrad TP, Lempicki RA, McLaughlin S, Pascuccio M, Gopaul R, McNally J, Cruz CC, Censoplano N, Chung E, Reitano KN, Kottilil S, Goode DJ, Fauci AS. (2008). "HIV-1 envelope protein binds to and signals through integrin alpha(4)beta(7), the gut mucosal homing receptor for peripheral T cells". Nature Immunol. In Press (3): 301. doi:10.1038/ni1566. PMID 18264102.
- ↑ 87.0 87.1 Pope M, Haase A (2003). "Transmission, acute HIV-1 infection and the quest for strategies to prevent infection". Nat Med 9 (7): 847–52. doi:10.1038/nm0703-847. PMID 12835704.
- ↑ doi:10.1073/pnas.0900502106
This citation will be automatically completed in the next few minutes. You can jump the queue or expand by hand - ↑ 89.0 89.1 89.2 Zheng, Y. H., Lovsin, N. and Peterlin, B. M. (2005). "Newly identified host factors modulate HIV replication". Immunol. Lett. 97 (2): 225–34. doi:10.1016/j.imlet.2004.11.026. PMID 15752562.
- ↑ Doc Kaiser's Microbiology Home Page > IV. VIRUSES > F. ANIMAL VIRUS LIFE CYCLES > 3. The Life Cycle of HIV Community College of Baltimore County. Updated: Jan., 2008
- ↑ Hiscott J, Kwon H, Genin P. (2001). "Hostile takeovers: viral appropriation of the NF-kappaB pathway". J Clin Invest. 107 (2): 143–151. doi:10.1172/JCI11918. PMID 11160127.
- ↑ Pollard, V. W. and Malim, M. H. (1998). "The HIV-1 Rev protein". Annu. Rev. Microbiol. 52: 491–532. doi:10.1146/annurev.micro.52.1.491. PMID 9891806.
- ↑ Gelderblom, H. R (1997). "Fine structure of HIV and SIV". In Los Alamos National Laboratory (ed.) (PDF). HIV Sequence Compendium. Los Alamos, New Mexico: Los Alamos National Laboratory. pp. 31–44. http://www.hiv.lanl.gov/content/sequence/HIV/COMPENDIUM/1997/partIII/Gelderblom.pdf.
- ↑ 94.0 94.1 94.2 94.3 Robertson DL, Hahn BH, Sharp PM. (1995). "Recombination in AIDS viruses". J Mol Evol. 40 (3): 249–59. doi:10.1007/BF00163230. PMID 7723052.
- ↑ Rambaut, A et al; Posada, D; Crandall, KA; Holmes, EC (January 2004). "The causes and consequences of HIV evolution". Nature Reviews Genetics 5 (52-61): 52–61. doi:10.1038/nrg1246. PMID 14708016. http://tree.bio.ed.ac.uk/downloadPaper.php?id=242.
- ↑ Perelson AS, Ribeiro RM (October 2008). "Estimating drug efficacy and viral dynamic parameters: HIV and HCV". Stat Med 27 (23): 4647–57. doi:10.1002/sim.3116. PMID 17960579.
- ↑ 97.0 97.1 PMID 19661993 (PubMed)
Citation will be completed automatically in a few minutes. Jump the queue or expand by hand - ↑ Holzammer S, Holznagel E, Kaul A, Kurth R, Norley S (2001). "High virus loads in naturally and experimentally SIVagm-infected African green monkeys". Virology 283 (2): 324–31. doi:10.1006/viro.2001.0870. PMID 11336557.
- ↑ Kurth, R. and Norley, S. (1996) Why don't the natural hosts of SIV develop simian AIDS?, J. NIH Res. 8, 33-37.
- ↑ Baier M, Dittmar MT, Cichutek K, Kurth R (1991). "Development of vivo of genetic variability of simian immunodeficiency virus". Proc. Natl. Acad. Sci. U.S.A. 88 (18): 8126–30. doi:10.1073/pnas.88.18.8126. PMID 1896460.
- ↑ Daniel MD, King NW, Letvin NL, Hunt RD, Sehgal PK, Desrosiers RC (1984). "A new type D retrovirus isolated from macaques with an immunodeficiency syndrome". Science 223 (4636): 602–5. doi:10.1126/science.6695172. PMID 6695172.
- ↑ 102.0 102.1 PMID 19626114 (PubMed)
Citation will be completed automatically in a few minutes. Jump the queue or expand by hand - ↑ Thomson, M. M., Perez-Alvarez, L. and Najera, R. (2002). "Molecular epidemiology of HIV-1 genetic forms and its significance for vaccine development and therapy". Lancet Infect. Dis. 2 (8): 461–471. doi:10.1016/S1473-3099(02)00343-2. PMID 12150845.
- ↑ Carr, J. K.; Foley, B. T., Leitner, T., Salminen, M., Korber, B. and McCutchan, F. (1998). "Reference Sequences Representing the Principal Genetic Diversity of HIV-1 in the Pandemic". In Los Alamos National Laboratory (ed.) (PDF). HIV Sequence Compendium. Los Alamos, New Mexico: Los Alamos National Laboratory. pp. 10–19. http://www.hiv.lanl.gov/content/sequence/HIV/COMPENDIUM/1998/III/Carr.pdf.
- ↑ Osmanov S, Pattou C, Walker N, Schwardlander B, Esparza J; WHO-UNAIDS Network for HIV Isolation and Characterization. (2002). "Estimated global distribution and regional spread of HIV-1 genetic subtypes in the year 2000". Acquir. Immune. Defic. Syndr. 29 (2): 184–190. PMID 11832690.
- ↑ Perrin L, Kaiser L, Yerly S. (2003). "Travel and the spread of HIV-1 genetic variants". Lancet Infect Dis. 3 (1): 22–27. doi:10.1016/S1473-3099(03)00484-5. PMID 12505029.
- ↑ 107.0 107.1 Plantier JC, Leoz M, Dickerson JE, et al. (August 2009). "A new human immunodeficiency virus derived from gorillas". Nat. Med. 15 (8): 871–2. doi:10.1038/nm.2016. PMID 19648927.
- ↑ Smith, L (2009-08-03). "Woman found carrying new strain of HIV from gorillas". London: The Independent. http://www.independent.co.uk/life-style/health-and-families/health-news/woman-found-carrying-new-strain-of-hiv-from-gorillas-1766627.html. Retrieved 2009-08-04.
- ↑ "Gorillas may be a source of AIDS, researchers find". Reuters India. 2009-08-03. http://in.reuters.com/article/worldNews/idINIndia-41490620090803. Retrieved 2009-08-04.
- ↑ 110.0 110.1 110.2 110.3 Kumaranayake, L. and Watts, C. (2001). "Resource allocation and priority setting of HIV/AIDS interventions: addressing the generalized epidemic in sub-Saharan Africa". J. Int. Dev. 13 (4): 451–466. doi:10.1002/jid.797.
- ↑ Kleinman, S. (September 2004). "Patient information: Blood donation and transfusion". Uptodate. http://www.uptodate.com/patients/content/topic.do?topicKey=blod_dis/2419. Retrieved 2008-04-03.
- ↑ 112.0 112.1 Centers for Disease Control and Prevention. (2001). "Revised guidelines for HIV counseling, testing, and referral". MMWR Recomm Rep. 50 (RR-19): 1–57. PMID 11718472.
- ↑ Celum CL, Coombs RW, Lafferty W, Inui TS, Louie PH, Gates CA, McCreedy BJ, Egan R, Grove T, Alexander S, et al. (1991). "Indeterminate human immunodeficiency virus type 1 western blots: seroconversion risk, specificity of supplemental tests, and an algorithm for evaluation". J Infect Dis. 164 (4): 656–664. PMID 1894929.
- ↑ Chou R, Huffman LH, Fu R, Smits AK, Korthuis PT (July 2005). "Screening for HIV: a review of the evidence for the U.S. Preventive Services Task Force". Ann. Intern. Med. 143 (1): 55–73. PMID 15998755. http://www.annals.org/cgi/content/full/143/1/55.
- ↑ Kleinman S, Busch MP, Hall L, et al. for the Retrovirus Epidemiology Donor Study (1998). "False-positive HIV-1 test results in a low-risk screening setting of voluntary blood donation". JAMA 280 (12): 1080–1085. doi:10.1001/jama.280.12.1080. PMID 9757856.
- ↑ Wood RW, Dunphy C, Okita K, Swenson P (2003). "Two "HIV-Infected" Persons Not Really Infected". Arch Intern Med 163 (15): 1857–1859. doi:10.1001/archinte.163.15.1857. PMID 12912724.
- ↑ Padeh YC, Rubinstein A, Shliozberg J (2005). "Common variable immunodeficiency and testing for HIV-1". N Engl J Med 353 (10): 1074–1075. doi:10.1056/NEJMc051339. PMID 16148302.
- ↑ Los Alamos National Laboratory • Established 1943. "Fighting the world’s most dangerous disease::Los Alamos Lab". Lanl.gov. http://www.lanl.gov/discover/curing_aids. Retrieved 2010-07-28.
- ↑ Robb ML (2008). "Failure of the Merck HIV vaccine: an uncertain step forward". Lancet 372 (9653): 1857–1858. doi:10.1016/S0140-6736(08)61593-7. PMID 19012958.
- ↑ "HIV vaccine 'reduces infection'". BBC News. September 24, 2009. http://news.bbc.co.uk/2/hi/health/8272113.stm. Retrieved March 30, 2010.
- ↑ Fan, H., Conner, R. F. and Villarreal, L. P. eds, ed (2005). AIDS : science and society (4th ed.). Boston, MA: Jones and Bartlett Publishers. ISBN 0-7637-0086-X.
- ↑ Karim, Q. A., Karim, S. S. A., Frolich, J. A., Grobler, A. C., Baxter, C., Mansoor, L. E., Kharsany, A. B. M., Sibeko, S., Mlisana, K. P., Omar, Z., Gengiah, T. N., Maarschalk, S., Arulappan, N., Mlotshwa, M., Morris, L., and Taylor, D. (19 July 2010). "Effectiveness and Safety of Tenofovir Gel, an Antiretroviral Microbicide, for the Prevention of HIV Infection in Women". Science. doi:10.1126/science.1193748. PMID 20643915.
- ↑ Department of Health and Human Services (January 2005). "A Pocket Guide to Adult HIV/AIDS Treatment January 2005 edition". http://www.hab.hrsa.gov/tools/HIVpocketguide05/PktGARTtables.htm. Retrieved 2006-01-17.
- ↑ 124.0 124.1 124.2 Palella, F. J., Delaney, K. M., Moorman, A. C., Loveless, M. O., Fuhrer, J., Satten, G. A., Aschman, D. J. and Holmberg, S. D. (1998). "Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection". N. Engl. J. Med. 338 (13): 853–860. doi:10.1056/NEJM199803263381301. PMID 9516219.
- ↑ Department of Health and Human Services Working Group on Antiretroviral Therapy and Medical Management of HIV-Infected Children (November 3, 2005). "Guidelines for the Use of Antiretroviral Agents in Pediatric HIV Infection" (PDF). http://www.aidsinfo.nih.gov/ContentFiles/PediatricGuidelines_PDA.pdf. Retrieved 2006-01-17.
- ↑ Department of Health and Human Services Panel on Clinical Practices for Treatment of HIV Infection (October 6, 2005). "Guidelines for the Use of Antiretroviral Agents in HIV-1-Infected Adults and Adolescents" (PDF). http://aidsinfo.nih.gov/ContentFiles/AdultandAdolescentGL.pdf. Retrieved 2006-01-17.
- ↑ Martinez-Picado, J., DePasquale, M. P., Kartsonis, N., Hanna, G. J., Wong, J., Finzi, D., Rosenberg, E., Gunthard, H.F., Sutton, L., Savara, A., Petropoulos, C. J., Hellmann, N., Walker, B. D., Richman, D. D., Siliciano, R. and D'Aquila, R. T. (2000). "Antiretroviral resistance during successful therapy of human immunodeficiency virus type 1 infection". Proc. Natl. Acad. Sci. U. S. A. 97 (20): 10948–10953. doi:10.1073/pnas.97.20.10948. PMID 11005867.
- ↑ Dybul, M., Fauci, A. S., Bartlett, J. G., Kaplan, J. E., Pau, A. K.; Panel on Clinical Practices for Treatment of HIV. (2002). "Guidelines for using antiretroviral agents among HIV-infected adults and adolescents". Ann. Intern. Med. 137 (5 Pt 2): 381–433. PMID 12617573.
- ↑ Blankson, J. N., Persaud, D., Siliciano, R. F. (2002). "The challenge of viral reservoirs in HIV-1 infection". Annu. Rev. Med. 53: 557–593. doi:10.1146/annurev.med.53.082901.104024. PMID 11818490.
- ↑ 130.0 130.1 Wood, E., Hogg, R. S., Yip, B., Harrigan, P. R., O'Shaughnessy, M. V. and Montaner, J. S. (2003). "Is there a baseline CD4 cell count that precludes a survival response to modern antiretroviral therapy?". AIDS 17 (5): 711–720. doi:10.1097/01.aids.0000050854.71999.d8 (inactive 2010-07-07). PMID 12646794.
- ↑ Chene, G., Sterne, J. A., May, M., Costagliola, D., Ledergerber, B., Phillips, A. N., Dabis, F., Lundgren, J., D'Arminio Monforte, A., de Wolf, F., Hogg, R., Reiss, P., Justice, A., Leport, C., Staszewski, S., Gill, J., Fatkenheuer, G., Egger, M. E. and the Antiretroviral Therapy Cohort Collaboration. (2003). "Prognostic importance of initial response in HIV-1 infected patients starting potent antiretroviral therapy: analysis of prospective studies". Lancet 362 (9385): 679–686. doi:10.1016/S0140-6736(03)14229-8. PMID 12957089.
- ↑ A computer based study in 2006, following the 2004 United States treatment guidelines: Schackman BR, Gebo KA, Walensky RP, Losina E, Muccio T, Sax PE, Weinstein MC, Seage GR 3rd, Moore RD, Freedberg KA. (2006). "The lifetime cost of current HIV care in the United States". Med Care 44 (11): 990–997. doi:10.1097/01.mlr.0000228021.89490.2a. PMID 17063130.
- ↑ Becker SL, Dezii CM, Burtcel B, Kawabata H, Hodder S. (2002). "Young HIV-infected adults are at greater risk for medication nonadherence". MedGenMed. 4 (3): 21. PMID 12466764.
- ↑ Nieuwkerk, P., Sprangers, M., Burger, D., Hoetelmans, R. M., Hugen, P. W., Danner, S. A., van Der Ende, M. E., Schneider, M. M., Schrey, G., Meenhorst, P. L., Sprenger, H. G., Kauffmann, R. H., Jambroes, M., Chesney, M. A., de Wolf, F., Lange, J. M. and the ATHENA Project. (2001). "Limited Patient Adherence to Highly Active Antiretroviral Therapy for HIV-1 Infection in an Observational Cohort Study". Arch. Intern. Med. 161 (16): 1962–1968. doi:10.1001/archinte.161.16.1962. PMID 11525698.
- ↑ Kleeberger, C., Phair, J., Strathdee, S., Detels, R., Kingsley, L. and Jacobson, L. P. (2001). "Determinants of Heterogeneous Adherence to HIV-Antiretroviral Therapies in the Multicenter AIDS Cohort Study". J. Acquir. Immune Defic. Syndr. 26 (1): 82–92. PMID 11176272.
- ↑ Heath, K. V., Singer, J., O'Shaughnessy, M. V., Montaner, J. S. and Hogg, R. S. (2002). "Intentional Nonadherence Due to Adverse Symptoms Associated With Antiretroviral Therapy". J. Acquir. Immune Defic. Syndr. 31 (2): 211–217. PMID 12394800.
- ↑ Montessori, V., Press, N., Harris, M., Akagi, L., Montaner, J. S. (2004). "Adverse effects of antiretroviral therapy for HIV infection.". CMAJ 170 (2): 229–238. PMID 14734438.
- ↑ Saitoh, A., Hull, A. D., Franklin, P. and Spector, S. A. (2005). "Myelomeningocele in an infant with intrauterine exposure to efavirenz". J. Perinatol. 25 (8): 555–556. doi:10.1038/sj.jp.7211343. PMID 16047034.
- ↑ Wang C, Vlahov D, Galai N, et al. (2004). "Mortality in HIV-seropositive versus seronegative persons in the era of highly active antiretroviral therapy.". J. Infect. Dis. 190 (6): 1046–54. doi:10.1086/422848. PMID 15319852.
- ↑ World Health Organisation (2006). "WHO case definitions of HIV for surveillance and revised clinical staging and immunological classification" (PDF). http://www.who.int/hiv/pub/guidelines/WHO%20HIV%20Staging.pdf. Retrieved 2006-12-27.
- ↑ 141.0 141.1 141.2 Ferrantelli F, Cafaro A, Ensoli B (December 2004). "Nonstructural HIV proteins as targets for prophylactic or therapeutic vaccines". Curr. Opin. Biotechnol. 15 (6): 543–56. doi:10.1016/j.copbio.2004.10.008. PMID 15560981.
- ↑ Mark Schoofs (2008). "A Doctor, a Mutation and a Potential Cure for AIDS". The Wall Street Journal. http://online.wsj.com/article/SB122602394113507555.html. Retrieved 2008-11-09.
- ↑ Hütter G, Nowak D, Mossner M, Ganepola S, Ganepola A, Allers K, Schneider T, Hofmann J, Kücherer C, Blau O, Blau IW, Hofmann WK, Thiel E (2009). "Long-Term Control of HIV by CCR5 Delta32/Delta32 Stem-Cell Transplantation". N Engl J Med 360 (7): 692–698. doi:10.1056/NEJMoa0802905. PMID 19213682. http://content.nejm.org/cgi/content/abstract/360/7/692. Retrieved 2009-03-31.
- ↑ Levy JA (2009). "Not an HIV Cure, but Encouraging New Directions". N Engl J Med 360 (7): 724–725. doi:10.1056/NEJMe0810248. PMID 19213687. http://content.nejm.org/cgi/content/full/360/7/724. Retrieved 2009-03-31.
- ↑ Glass WG, McDermott DH, Lim JK, et al. (January 2006). "CCR5 deficiency increases risk of symptomatic West Nile virus infection". J. Exp. Med. 203 (1): 35–40. doi:10.1084/jem.20051970. PMID 16418398.
- ↑ 146.0 146.1 Swanson MD, Winter HC, Goldstein IJ, Markovitz DM (March 2010). "A Lectin Isolated from Bananas Is a Potent Inhibitor of HIV Replication". J. Biol. Chem. 285 (12): 8646–55. doi:10.1074/jbc.M109.034926. PMID 20080975.
- ↑ 147.0 147.1 "Study: Chemical in Bananas Could Help Fight HIV", FOX News, March 16, 2010
- ↑ "Protein in bananas could help block spread of HIV, University of Michigan researchers say", AnnArbor.com, March 15, 2010
- ↑ Archin NM, Eron JJ, Palmer S, Hartmann-Duff A, Martinson JA, Wiegand A, Bandarenko N, Schmitz JL, Bosch RJ, Landay AL, Coffin JM, Margolis DM. (2008). "Valproic acid without intensified antiviral therapy has limited impact on persistent HIV infection of resting CD4+ T cells.". AIDS 22 (10): 1131–1135. doi:10.1097/QAD.0b013e3282fd6df4. PMID 18525258.
- ↑ Bowman MC, Archin NM, Margolis DM. (2009). "Pharmaceutical approaches to eradication of persistent HIV infection.". Expert Reviews in Molecular Medicine 11 (e6): e6. doi:10.1017/S1462399409000970. PMID 19208267.
- ↑ 151.0 151.1 UNAIDS, WHO (December 2007). "2007 AIDS epidemic update" (PDF). http://data.unaids.org/pub/EPISlides/2007/2007_epiupdate_en.pdf. Retrieved 2008-03-12.
- ↑ Zwahlen M, Egger M (2006) (PDF). Progression and mortality of untreated HIV-positive individuals living in resource-limited settings: update of literature review and evidence synthesis. UNAIDS Obligation HQ/05/422204. http://data.unaids.org/pub/Periodical/2006/zwahlen_unaids_hq_05_422204_2007_en.pdf. Retrieved 2008-03-19.
- ↑ Knoll B, Lassmann B, Temesgen Z (2007). "Current status of HIV infection: a review for non-HIV-treating physicians". Int J Dermatol 46 (12): 1219–28. doi:10.1111/j.1365-4632.2007.03520.x (inactive 2009-08-10). PMID 18173512.
- ↑ Antiretroviral Therapy Cohort Collaboration (2008). "Life expectancy of individuals on combination antiretroviral therapy in high-income countries: a collaborative analysis of 14 cohort studies". Lancet 372 (9635): 293–9. doi:10.1016/S0140-6736(08)61113-7. PMID 18657708.
- ↑ Clerici M, Balotta C, Meroni L, et al. (1996). "Type 1 cytokine production and low prevalence of viral isolation correlate with long-term non progression in HIV infection". AIDS Res. Hum. Retroviruses. 12 (11): 1053–1061. doi:10.1089/aid.1996.12.1053. PMID 8827221.
- ↑ Morgan D, Mahe C, Mayanja B, Whitworth JA (2002). "Progression to symptomatic disease in people infected with HIV-1 in rural Uganda: prospective cohort study". BMJ 324 (7331): 193–196. doi:10.1136/bmj.324.7331.193. PMID 11809639.
- ↑ Campbell GR, Pasquier E, Watkins J, et al. (2004). "The glutamine-rich region of the HIV-1 Tat protein is involved in T-cell apoptosis". J. Biol. Chem. 279 (46): 48197–48204. doi:10.1074/jbc.M406195200. PMID 15331610.
- ↑ Campbell GR, Watkins JD, Esquieu D, Pasquier E, Loret EP, Spector SA (2005). "The C terminus of HIV-1 Tat modulates the extent of CD178-mediated apoptosis of T cells". J. Biol. Chem. 280 (46): 38376–39382. doi:10.1074/jbc.M506630200. PMID 16155003.
- ↑ Senkaali D, Muwonge R, Morgan D, Yirrell D, Whitworth J, Kaleebu P (2005). "The relationship between HIV type 1 disease progression and V3 serotype in a rural Ugandan cohort". AIDS Res. Hum. Retroviruses. 20 (9): 932–937. doi:10.1089/aid.2004.20.932. PMID 15585080.
- ↑ "UN Millenium Goals report 2010". United Nations, www.un.org. http://www.un.org/millenniumgoals/pdf/MDG%20Report%202010%20En%20r15%20-low%20res%2020100615%20-.pdf. Retrieved 2010-07-03.
- ↑ McNeil, Jr., Donald (November 20, 2007). "U.N. Agency to Say It Overstated Extent of H.I.V. Cases by Millions". The New York Times. http://query.nytimes.com/gst/fullpage.html?res=9C01EEDF103BF933A15752C1A9619C8B63&n. Retrieved 2008-01-16.
- ↑ Hakim, James (August 2009). HIV/AIDS: an update on Epidemiology, Prevention, and Treatment. [Southern Sudan Medical Journal].
- ↑ UNAIDS (2001). "Special Session of the General Assembly on HIV/AIDS Round table 3 Socio-economic impact of the epidemic and the strengthening of national capacities to combat HIV/AIDS" (PDF). http://data.unaids.org/Publications/External-Documents/GAS26-rt3_en.pdf. Retrieved 2006-06-15.
- ↑ World Bank (2005). "Evaluating the World Bank's Assistance for Fighting the HIV/AIDS Epidemic". http://www.worldbank.org/oed/aids/main_report.html. Retrieved 2006-01-17.
- ↑ Worobey M, Gemmel M, Teuwen DE, et al. (October 2008). "Direct evidence of extensive diversity of HIV-1 in Kinshasa by 1960". Nature 455 (7213): 661–4. doi:10.1038/nature07390. PMID 18833279.
- ↑ "Pneumocystis Pneumonia -- Los Angeles". http://www.cdc.gov/MMWR/preview/mmwrhtml/00043494.htm. Retrieved 2008-05-05.
- ↑ Gao F, Bailes E, Robertson DL, et al. (February 1999). "Origin of HIV-1 in the chimpanzee Pan troglodytes troglodytes". Nature 397 (6718): 436–41. doi:10.1038/17130. PMID 9989410.
- ↑ Keele, B. F., van Heuverswyn, F., Li, Y. Y., Bailes, E., Takehisa, J., Santiago, M. L., Bibollet-Ruche, F., Chen, Y., Wain, L. V., Liegois, F., Loul, S., Mpoudi Ngole, E., Bienvenue, Y., Delaporte, E., Brookfield, J. F. Y., Sharp, P. M., Shaw, G. M., Peeters, M., and Hahn, B. H. (28 July 2006). "Chimpanzee Reservoirs of Pandemic and Nonpandemic HIV-1". Science 313 (5786 pages=523–526): 523–6. doi:10.1126/science.1126531. PMID 16728595.
- ↑ Goodier, J., and Kazazian, H. (2008). "Retrotransposons Revisited: The Restraint and Rehabilitation of Parasites". Cell 135 (1): 23–35. doi:10.1016/j.cell.2008.09.022. PMID 18854152.
- ↑ Centers for Disease Control (CDC) (June 1982). "A cluster of Kaposi's sarcoma and Pneumocystis carinii pneumonia among homosexual male residents of Los Angeles and Orange Counties, California". MMWR Morb. Mortal. Wkly. Rep. 31 (23): 305–7. PMID 6811844. http://www.cdc.gov/mmwr/preview/mmwrhtml/00001114.htm.
- ↑ Centers for Disease Control (CDC) (June 1981). "Pneumocystis pneumonia--Los Angeles". MMWR Morb. Mortal. Wkly. Rep. 30 (21): 250–2. PMID 6265753.
- ↑ Masur H, Michelis MA, Greene JB, et al. (December 1981). "An outbreak of community-acquired Pneumocystis carinii pneumonia: initial manifestation of cellular immune dysfunction". N. Engl. J. Med. 305 (24): 1431–8. doi:10.1056/NEJM198112103052402. PMID 6975437.
- ↑ Gottlieb MS, Schroff R, Schanker HM, et al. (December 1981). "Pneumocystis carinii pneumonia and mucosal candidiasis in previously healthy homosexual men: evidence of a new acquired cellular immunodeficiency". N. Engl. J. Med. 305 (24): 1425–31. doi:10.1056/NEJM198112103052401. PMID 6272109. http://www.nejm.org/doi/abs/10.1056/NEJM198112103052401.
- ↑ Friedman-Kien AE (October 1981). "Disseminated Kaposi's sarcoma syndrome in young homosexual men". J. Am. Acad. Dermatol. 5 (4): 468–71. doi:10.1016/S0190-9622(81)80010-2. PMID 7287964.
- ↑ Hymes KB, Cheung T, Greene JB, et al. (September 1981). "Kaposi's sarcoma in homosexual men-a report of eight cases". Lancet 2 (8247): 598–600. PMID 6116083.
- ↑ 176.0 176.1 Basavapathruni, A; Anderson, KS (December 2007). "Reverse transcription of the HIV-1 pandemic". The FASEB Journal 21 (14): 3795–3808. doi:10.1096/fj.07-8697rev. PMID 17639073.
- ↑ RC Gallo, PS Sarin, EP Gelmann, M Robert-Guroff, E Richardson, VS Kalyanaraman, D Mann, GD Sidhu, RE Stahl, S Zolla-Pazner, J Leibowitch, and M Popovic (1983). "Isolation of human T-cell leukemia virus in acquired immune deficiency syndrome (AIDS)". Science 220 (4599): 865–867. doi:10.1126/science.6189183. PMID 6601823.
- ↑ F Barre-Sinoussi, JC Chermann, F Rey, MT Nugeyre, S Chamaret, J Gruest, C Dauguet, C Axler-Blin, F Vezinet-Brun, C Rouzioux, W Rozenbaum, and L Montagnier (1983). "Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS)". Science 220: 868–870. doi:10.1126/science.6601823.
- ↑ "The Nobel Prize in Physiology or Medicine 2008". Nobel Foundation. http://nobelprize.org/nobel_prizes/medicine/laureates/2008/index.html. Retrieved October 28, 2009.
- ↑ 180.0 180.1 Cohen, J; Enserink, Martin (10 October 2008). "Nobel Prize in Physiology or Medicine: HIV, HPV researchers honored, but one scientist is left out". Science 322 (5899): 149–175. doi:10.1126/science.322.5899.174. PMID 18845715.
- ↑ Altman, Lawrence (2008-10-06). "Three Europeans Win the 2008 Nobel for Medicine". New York Times. http://www.nytimes.com/2008/10/07/health/07nobel.html. Retrieved 2008-10-06.
- ↑ 182.0 182.1 Kalichman, Seth (2009). Denying AIDS: Conspiracy Theories, Pseudoscience, and Human Tragedy. New York: Copernicus Books (Springer Science+Business Media). ISBN 978-0-387-79475-4. http://books.google.com/?id=_mtDBCDwxugC&printsec=frontcover&q=.
- ↑ Duesberg, P. H. (1988). "HIV is not the cause of AIDS". Science 241 (4865): 514, 517. doi:10.1126/science.3399880. PMID 3399880. Cohen, J. (1994). "The Controversy over HIV and AIDS" (PDF). Science 266 (5191): 1642–1649. doi:10.1126/science.7992043. PMID 7992043. http://www.sciencemag.org/feature/data/cohen/266-5191-1642a.pdf. Retrieved 2009-03-31.
- ↑ Smith TC, Novella SP (August 2007). "HIV denial in the Internet era". PLoS Med. 4 (8): e256. doi:10.1371/journal.pmed.0040256. PMID 17713982. PMC 1949841. http://medicine.plosjournals.org/perlserv/?request=get-document&doi=10.1371/journal.pmed.0040256&ct=1&SESSID=3d4baa1a64e57d8ff33e9d41eb2335a1. Retrieved 2009-11-07.
- ↑
- "The Durban Declaration". Nature 406 (6791): 15–6. 2000. doi:10.1038/35017662. PMID 10894520..
- Various. "Resources and Links, HIV-AIDS Connection". National Institute of Allergy and Infectious Diseases. http://www3.niaid.nih.gov/topics/HIVAIDS/Understanding/How+HIV+Causes+AIDS/connection.htm. Retrieved 2009-02-22.
- O'Brien SJ, Goedert JJ (1996). "HIV causes AIDS: Koch's postulates fulfilled". Curr. Opin. Immunol. 8 (5): 613–8. doi:10.1016/S0952-7915(96)80075-6. PMID 8902385.
- Galéa P, Chermann JC (1998). "HIV as the cause of AIDS and associated diseases". Genetica 104 (2): 133–42. doi:10.1023/A:1003432603348. PMID 10220906.
- ↑ Watson J (2006). "Scientists, activists sue South Africa's AIDS 'denialists'". Nat. Med. 12 (1): 6. doi:10.1038/nm0106-6a. PMID 16397537.
- ↑ Baleta A (2003). "S Africa's AIDS activists accuse government of murder". Lancet 361 (9363): 1105. doi:10.1016/S0140-6736(03)12909-1. PMID 12672319.
- ↑ Cohen J (2000). "South Africa's new enemy". Science 288 (5474): 2168–70. doi:10.1126/science.288.5474.2168. PMID 10896606.
External links
- HIV/AIDS at the Open Directory Project
- "AIDSinfo - HIV/AIDS Treatment Information". US Department of Health and Human Services. http://aidsinfo.nih.gov/. Retrieved 2008-03-21.
- "UNAIDS: The Joint United Nations Programme on HIV/AIDS". UNAIDS. http://www.unaids.org/en/. Retrieved 2008-03-21.
- "AIDS.gov: Portal to all Federal HIV/AIDS information". AIDS.gov. http://www.AIDS.gov. Retrieved 2008-05-20.
- "HIV News and Resources". The Body. http://www.thebody.com/. Retrieved 2008-07-18.
- American Psychological Association's Office on AIDS
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||
|
|||||