Fibrin
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
Fibrin (also called Factor Ia) is a fibrous protein involved in the clotting of blood, and is non-globular. It is a fibrillar protein that is polymerised to form a "mesh" that forms a hemostatic plug or clot (in conjunction with platelets) over a wound site.
Fibrin is made from fibrinogen, a soluble plasma glycoprotein that is synthesised by the liver. Processes in the coagulation cascade activate the zymogen prothrombin to the serine protease thrombin, which is responsible for converting fibrinogen into fibrin. Fibrin is then cross linked by factor XIII to form a clot. FXIIIa stabilizes fibrin further by incorporation of the fibrinolysis inhibitors alpha-2-antiplasmin and TAFI (thrombin activatable fibrinolysis inhibitor, procarboxypeptidase B), and binding to several adhesive proteins of various cells.[2] Both the activation of Factor XIII by thrombin and plasminogen activator (t-PA) are catalyzed by fibrin.[2] Fibrin specifically binds the activated coagulation factors factor Xa and thrombin and entraps them in the network of fibers, thus functioning as a temporary inhibitor of these enzymes which stay active and can be released during fibrinolysis.[3] Recent research has shown that fibrin plays a key role in the inflammatory response and development of rheumatoid arthritis.
Fibrin is involved in the following biological processes: signal transduction, blood coagulation, platelet activation, and protein polymerization.
Contents |
Role in disease
Excessive generation of fibrin due to activation of the coagulation cascade leads to thrombosis, more commonly known as a clot, while ineffective generation or premature lysis of fibrin predisposes to hemorrhage.
Dysfunction or disease of the liver can lead to a decrease in fibrinogen production or the production of abnormal fibrinogen molecules with reduced activity (dysfibrinogenaemia). Hereditary abnormalities of fibrinogen (the gene is carried on chromosome 4) are of both quantitative and qualitative in nature and include; afibrinogenaemia, hypofibrinogenaemia, dysfibrinogenaemia, and hypodysfibrinogenaemia.
Consequences of reduced, absent, or dysfunctional fibrin is likely to render patients as hemophiliacs.
Fibrinogen deficiency
Congenital deficiency (afibrinognenemia) or disturbed function of fibrinogen has been described in a few cases.[4]
It can lead to either bleeding, thromboembolic complications or is clinically without pathological findings. More common are acquired deficiency stages which can be detected by laboratory tests in blood plasma or in whole blood by means of thrombelastometry.[5] Acquired deficiency is found after hemodilution, blood losses and/or consumption such as in trauma patients, during some phases of disseminated intravascular coagulation (DIC), and also in sepsis. In patients with fibrinogen deficiency, the correction of bleeding is possible by infusion of fresh frozen plasma (FFP), cryoprecipitate (a fibrinogen rich plasma fraction) or by fibrinogen concentrates. There is increasing evidence that correction of fibrinogen deficiency or fibrinogen polymerization disorders is very important in patients with bleeding.[6]
Diagnostic use
Fibrinogen levels can be measured in venous blood. Normal levels are about 1.5-2.77 g/L, depending on the method which is used. Typically fibrinogen is measured in citrated plasma samples in the laboratory, however the analysis of whole blood samples by use of thrombelastometry (platelet function is inhibited with cytochalasin D) is also possible.[5] Higher levels are, amongst others, associated with cardiovascular disease (>3.43 g/L). It may be elevated in any form of inflammation, as it is an acute phase protein; for example, it is especially apparent in human gingival tissue during the initial phase of periodontal disease.[7]
It is used in veterinary medicine as an inflammatory marker: in horses a level above the normal range of 1.0-4.0 g/L suggests some degree of systemic inflammatory response.
Low levels of fibrinogen can indicate a systemic activation of the clotting system, with consumption of clotting factors faster than synthesis. This excessive clotting factor consumption condition is known as Disseminated Intravascular Coagulation or "DIC." DIC can be difficult to diagnose, but a strong clue is low fibrinogen levels in the setting of prolonged clotting times (PT or [[Partial thromboplastin time|aPTT), in the context of acute critical illness such as sepsis or trauma. Besides low fibrinogen level, fibrin polymerization disorders which can be induced by several factors, including plasma expanders, can also lead to severe bleeding problems.[5] Fibrin polymerization disorders can be detected by viscoelastic methods such as thrombelastometry.[5]
Physiology
Fibrinogen (also called factor I) is a 340 KDa glycoprotein synthesised in the liver by hepatocytes and megakaryocytes. The concentration in blood plasma is 1.5-4.0 g/L (normally measured using the Clauss method) or about 7 µM. In its natural form, fibrinogen can form bridges between platelets, by binding to their GpIIb/IIIa surface membrane proteins; however its major function is as the precursor to fibrin.
Fibrinogen, the principal protein of vertebrate blood clotting is a hexamer containing two sets of three different chains (α, β, and γ), linked to each other by disulfide bonds. The N-terminal sections of these three chains contain the cysteines that participate in the cross-linking of the chains. The C-terminal parts of the α, β and γ chains contain a domain of about 225 amino-acid residues, which can function as a molecular recognition unit. In fibrinogen as well as in angiopoietin this domain is implicated in protein-protein interactions. In lectins, such as mammalian ficolins and invertebrate tachylectin 5A, the fibrinogen C-terminal domain binds carbohydrates. On the fibrinogen α and β chains, there is a small peptide sequence (called a fibrinopeptide). These small peptides are what prevent fibrinogen from spontaneously forming polymers with itself.[8]
Fibrin from different animal sources is generally glycosylated with complex type diantennary asparagine linked glycans. Variety is just found in the degree of core fucosylation and in the type of sialic acid and galactose linkage.[9]
Structure
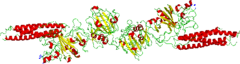
The image at the left is a crystal structure of the double-d fragment from human fibrin with two bound ligands. The experimental method used to obtain the image was X-ray diffraction, and it has a resolution of 2.30 Å. The structure is mainly made up of single alpha helices shown in red and beta sheets shown in yellow. The two blue structures are the bound ligands. The chemical structures of the ligands are Ca+2 ion, alpha-D-mannose (C6H12O6), and D-glucosamine (C8H15NO6).
See also
- Fibrin glue
- Fibrin Scaffold
References
- ↑ PDB 1FZC; Everse SJ, Spraggon G, Veerapandian L, Riley M, Doolittle RF (June 1998). "Crystal structure of fragment double-D from human fibrin with two different bound ligands". Biochemistry 37 (24): 8637–42. doi:10.1021/bi9804129. PMID 9628725.
- ↑ 2.0 2.1 Muszbek L, Bagoly Z, Bereczky Z, Katona E (July 2008). "The involvement of blood coagulation factor XIII in fibrinolysis and thrombosis". Cardiovascular & Hematological Agents in Medicinal Chemistry 6 (3): 190–205. doi:10.2174/187152508784871990. PMID 18673233.
- ↑ Kaiser B (2003). "DX-9065a, a direct inhibitor of factor Xa". Cardiovascular Drug Reviews 21 (2): 91–104. PMID 12847561.
- ↑ Acharya SS, Dimichele DM (November 2008). "Rare inherited disorders of fibrinogen". Haemophilia : the Official Journal of the World Federation of Hemophilia 14 (6): 1151–8. doi:10.1111/j.1365-2516.2008.01831.x (inactive 2009-05-14). PMID 19141154.
- ↑ 5.0 5.1 5.2 5.3 Lang T, Johanning K, Metzler H, Piepenbrock S, Solomon C, Rahe-Meyer N, Tanaka KA (March 2009). "The effects of fibrinogen levels on thromboelastometric variables in the presence of thrombocytopenia". Anesthesia and Analgesia 108 (3): 751–8. doi:10.1213/ane.0b013e3181966675. PMID 19224779.
- ↑ Fries D, Innerhofer P, Schobersberger W (April 2009). "Time for changing coagulation management in trauma-related massive bleeding". Current Opinion in Anaesthesiology 22 (2): 267–74. doi:10.1097/ACO.0b013e32832678d9 (inactive 2009-05-14). PMID 19390253.
- ↑ Page RC, Schroeder HE (March 1976). "Pathogenesis of inflammatory periodontal disease. A summary of current work". Lab. Invest. 34 (3): 235–49. PMID 765622.
- ↑ [1] Fibrinogen C-terminal domain in PROSITE
- ↑ Pabst M, Bondili JS, Stadlmann J, Mach L, Altmann F (July 2007). "Mass + retention time = structure: a strategy for the analysis of N-glycans by carbon LC-ESI-MS and its application to fibrin N-glycans". Anal. Chem. 79 (13): 5051–7. doi:10.1021/ac070363i. PMID 17539604.
External links
- TGW1916.net, Defibrinated blood harvested from sheep (video)
|
|||||||||||||||||||||||||||||||
|
||||||||||||||||||||