Tumor necrosis factor-alpha
| edit |
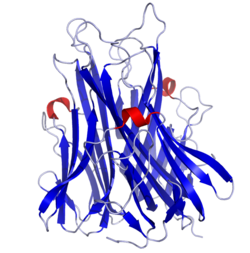
Tumor necrosis factor (TNF, cachexin or cachectin and formally known as tumor necrosis factor-alpha) is a cytokine involved in systemic inflammation and is a member of a group of cytokines that stimulate the acute phase reaction.
The primary role of TNF is in the regulation of immune cells. TNF is also able to induce apoptotic cell death, to induce inflammation, and to inhibit tumorigenesis and viral replication.
Dysregulation and, in particular, overproduction of TNF have been implicated in a variety of human diseases, as well as cancer.[1]
Contents |
Discovery
The theory of an anti-tumoral response of the immune system in vivo was recognized 100 years ago by the physician William B. Coley. In 1968, Dr. Gale A Granger from the University of California, Irvine, reported a cytotoxic factor produced by lymphocytes and named it lymphotoxin (LT).[2] Credit for this discovery is shared by Dr. Nancy H. Ruddle from Yale University, who reported the same activity in a series of back-to-back articles published in the same month and year.[3] Subsequently in 1975 Dr. Lloyd Old from Memorial Sloan-Kettering Cancer Center, New York, reported another cytotoxic factor produced by macrophages, and named it tumor necrosis factor (TNF).[4] Both factors were described based on their ability to kill mouse fibrosarcoma L-929 cells.
When the cDNAs encoding LT and TNF were cloned in 1984,[5] they were revealed to be similar. The binding of TNF to its receptor and its displacement by LT confirmed the functional homology between the two factors. The sequential and functional homology of TNF and LT led to the renaming of TNF as TNFα and LT as TNFβ. In 1985, Bruce A. Beutler and Anthony Cerami discovered that a hormone that induces cachexia and previously-named cachectin was actually TNF.[6] These investigators then identified TNF as the key mediator of septic shock in response to infection.[7] Subsequently, it was recognized that TNF is the prototypic member of a large cytokine family, the TNF family.
Gene
The human TNF gene (TNFA) was cloned in 1985.[8] It maps to chromosome 6p21.3, spans about 3 kb and contains 4 exons. The last exon codes for more than 80% of the secreted protein.[9] The 3' UTR of TNF alpha contains an AU-rich element (ARE).
Structure
TNF is primarily produced as a 212-amino acid-long type II transmembrane protein arranged in stable homotrimers.[10][11] From this membrane-integrated form the soluble homotrimeric cytokine (sTNF) is released via proteolytic cleavage by the metalloprotease TNF alpha converting enzyme (TACE, also called ADAM17).[12] The soluble 51 kDa trimeric sTNF tends to dissociate at concentrations below the nanomolar range, thereby losing its bioactivity.
The 17-kilodalton (kDa) TNF protomers (185-amino acid-long) are composed of two antiparallel β-pleated sheets with antiparallel β-strands, forming a 'jelly roll' β-structure, typical for the TNF family, but also found in viral capsid proteins.
Cell Signaling
Two receptors, TNF-R1 (TNF receptor type 1; CD120a; p55/60) and TNF-R2 (TNF receptor type 2; CD120b; p75/80), bind to TNF. TNF-R1 is expressed in most tissues, and can be fully activated by both the membrane-bound and soluble trimeric forms of TNF, whereas TNF-R2 is found only in cells of the immune system, and respond to the membrane-bound form of the TNF homotrimer. As most information regarding TNF signaling is derived from TNF-R1, the role of TNF-R2 is likely underestimated.

Upon contact with their ligand, TNF receptors also form trimers, their tips fitting into the grooves formed between TNF monomers. This binding causes a conformational change to occur in the receptor, leading to the dissociation of the inhibitory protein SODD from the intracellular death domain. This dissociation enables the adaptor protein TRADD to bind to the death domain, serving as a platform for subsequent protein binding. Following TRADD binding, three pathways can be initiated.[13][14]
- Activation of NF-kB: TRADD recruits TRAF2 and RIP. TRAF2 in turn recruits the multicomponent protein kinase IKK, enabling the serine-threonine kinase RIP to activate it. An inhibitory protein, IκBα, that normally binds to NF-κB and inhibits its translocation, is phosphorylated by IKK and subsequently degraded, releasing NF-κB. NF-κB is a heterodimeric transcription factor that translocates to the nucleus and mediates the transcription of a vast array of proteins involved in cell survival and proliferation, inflammatory response, and anti-apoptotic factors.
- Activation of the MAPK pathways: Of the three major MAPK cascades, TNF induces a strong activation of the stress-related JNK group, evokes moderate response of the p38-MAPK, and is responsible for minimal activation of the classical ERKs. TRAF2 activates the JNK-inducing upstream kinases of MEKK1 and ASK1 (either directly or through GCKs and Trx, respectively), and these two kinases phosphorylate MKK7, which then activates JNK. JNK translocates to the nucleus and activates transcription factors such as c-Jun and ATF2. The JNK pathway is involved in cell differentiation, proliferation, and is generally pro-apoptotic.
- Induction of death signaling: Like all death-domain-containing members of the TNFR superfamily, TNF-R1 is involved in death signaling.[15] However, TNF-induced cell death plays only a minor role compared to its overwhelming functions in the inflammatory process. Its death-inducing capability is weak compared to other family members (such as Fas), and often masked by the anti-apoptotic effects of NF-κB. Nevertheless, TRADD binds FADD, which then recruits the cysteine protease caspase-8. A high concentration of caspase-8 induces its autoproteolytic activation and subsequent cleaving of effector caspases, leading to cell apoptosis.
The myriad and often-conflicting effects mediated by the above pathways indicate the existence of extensive cross-talk. For instance, NF-κB enhances the transcription of C-FLIP, Bcl-2, and cIAP, inhibitory proteins that interfere with death signaling. On the other hand, activated caspases cleave several components of the NF-κB pathway, including RIP, IKK, and the subunits of NF-κB itself. Other factors, such as cell type, concurrent stimulation of other cytokines, or the amount of reactive oxygen species (ROS) can shift the balance in favor of one pathway or another. Such complicated signaling ensures that, whenever TNF is released, various cells with vastly diverse functions and conditions can all respond appropriately to inflammation.
Physiology
TNF is produced mainly by macrophages, but they are produced also by a broad variety of other cell types including lymphoid cells, mast cells, endothelial cells, cardiac myocytes, adipose tissue, fibroblasts, and neuronal tissue. Large amounts of TNF are released in response to lipopolysaccharide, other bacterial products, and Interleukin-1 (IL-1).
It has a number of actions on various organ systems, generally together with IL-1 and Interleukin-6 (IL-6):
- On the hypothalamus:
- Stimulating of the hypothalamic-pituitary-adrenal axis by stimulating the release of corticotropin releasing hormone (CRH)
- Suppressing appetite
- Fever
- On the liver: stimulating the acute phase response, leading to an increase in C-reactive protein and a number of other mediators. It also induces insulin resistance by promoting serine-phosphorylation of insulin receptor substrate-1 (IRS-1), which impairs insulin signaling
- It is a potent chemoattractant for neutrophils, and helps them to stick to the endothelial cells for migration
- On macrophages: stimulates phagocytosis, and production of IL-1 oxidants and the inflammatory lipid prostaglandin E2 PGE2
- On other tissues: increasing insulin resistance.
A local increase in concentration of TNF will cause the cardinal signs of Inflammation to occur: heat, swelling, redness, and pain.
Whereas high concentrations of TNF induce shock-like symptoms, the prolonged exposure to low concentrations of TNF can result in cachexia, a wasting syndrome. This can be found, for example, in tumor patients.
Pharmacology
Tumor necrosis factor promotes the inflammatory response, which, in turn, causes many of the clinical problems associated with autoimmune disorders such as rheumatoid arthritis, ankylosing spondylitis, Crohn's disease, psoriasis and refractory asthma. These disorders are sometimes treated by using a TNF inhibitor. This inhibition can be achieved with a monoclonal antibody such as infliximab (Remicade) or adalimumab (Humira), or with a circulating receptor fusion protein such as etanercept (Enbrel).
See also
- Lymphotoxin (Tumor necrosis factor-beta)
References
- ↑ Locksley RM, Killeen N, Lenardo MJ (2001). "The TNF and TNF receptor superfamilies: integrating mammalian biology". Cell 104 (4): 487–501. doi:. PMID 11239407.
- ↑ Kolb WP, Granger GA (1968). "Lymphocyte in vitro cytotoxicity: characterization of human lymphotoxin". Proc. Natl. Acad. Sci. U.S.A. 61 (4): 1250–5. doi:. PMID 5249808.
- ↑ Ruddle NH, Waksman BH (December 1968). "Cytotoxicity mediated by soluble antigen and lymphocytes in delayed hypersensitivity. 3. Analysis of mechanism". J. Exp. Med. 128 (6): 1267–79. PMID 5693925.
- ↑ Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B (1975). "An endotoxin-induced serum factor that causes necrosis of tumors". Proc. Natl. Acad. Sci. U.S.A. 72 (9): 3666–70. doi:. PMID 1103152.
- ↑ Pennica D, Nedwin GE, Hayflick JS, Seeburg PH, Derynck R, Palladino MA, Kohr WJ, Aggarwal BB, Goeddel DV (1984). "Human tumour necrosis factor: precursor structure, expression and homology to lymphotoxin". Nature 312 (5996): 724–9. doi:. PMID 6392892.
- ↑ Beutler B, Greenwald D, Hulmes JD, Chang M, Pan YC, Mathison J, Ulevitch R, Cerami A (1985). "Identity of tumour necrosis factor and the macrophage-secreted factor cachectin". Nature 316 (6028): 552–4. doi:. PMID 2993897.
- ↑ Beutler B, Milsark IW, Cerami AC (August 1985). "Passive immunization against cachectin/tumor necrosis factor protects mice from lethal effect of endotoxin". Science (journal) 229 (4716): 869–71. doi:. PMID 3895437.
- ↑ Old LJ (1985). "Tumor necrosis factor (TNF)". Science 230 (4726): 630–2. doi:. PMID 2413547.
- ↑ Nedwin GE, Naylor SL, Sakaguchi AY, Smith D, Jarrett-Nedwin J, Pennica D, Goeddel DV, Gray PW (1985). "Human lymphotoxin and tumor necrosis factor genes: structure, homology and chromosomal localization". Nucleic Acids Res. 13 (17): 6361–73. doi:. PMID 2995927. http://www.pubmedcentral.nih.gov/picrender.fcgi?artid=321958&blobtype=pdf.
- ↑ Kriegler M, Perez C, DeFay K, Albert I, Lu SD (1988). "A novel form of TNF/cachectin is a cell surface cytotoxic transmembrane protein: ramifications for the complex physiology of TNF". Cell 53 (1): 45–53. doi:. PMID 3349526.
- ↑ Tang P, Hung M-C, Klostergaard J (1996). "Human pro-tumor necrosis factor is a homotrimer". Biochemistry 35 (25): 8216–25. doi:. PMID 8679576.
- ↑ Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, Castner BJ, Stocking KL, Reddy P, Srinivasan S, Nelson N, Boiani N, Schooley KA, Gerhart M, Davis R, Fitzner JN, Johnson RS, Paxton RJ, March CJ, Cerretti DP (1997). "A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells". Nature 385 (6618): 729–33. doi:. PMID 9034190.
- ↑ Wajant H, Pfizenmaier K, Scheurich P (2003). "Tumor necrosis factor signaling". Cell Death Differ. 10 (1): 45–65. doi:. PMID 12655295.
- ↑ Chen G, Goeddel DV (2002). "TNF-R1 signaling: a beautiful pathway". Science 296 (5573): 1634–5. doi:. PMID 12040173.
- ↑ Gaur U, Aggarwal BB (2003). "Regulation of proliferation, survival and apoptosis by members of the TNF superfamily". Biochem. Pharmacol. 66 (8): 1403–8. doi:. PMID 14555214.
External links
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||