Specific heat capacity
| Material Properties | |
|---|---|
| Specific heat | 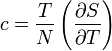 |
| Compressibility | 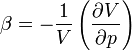 |
| Thermal expansion | 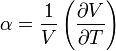 |
Specific heat capacity, also known simply as specific heat, is the measure of the heat energy required to increase the temperature of a unit quantity of a substance by a certain temperature interval. The term originated primarily through the work of Scottish physicist Joseph Black who conducted various heat measurements and used the phrase “capacity for heat.”[1] More heat energy is required to increase the temperature of a substance with high specific heat capacity than one with low specific heat capacity. For instance, eight times the heat energy is required to increase the temperature of an ingot of magnesium as is required for a lead ingot of the same mass. The specific heat of virtually any substance can be measured, including chemical elements, compounds, alloys, solutions, and composites.
The symbols for specific heat capacity are either C or c depending on how the quantity of a substance is measured (see Symbols and standards below for usage rules). In the measurement of physical properties, the term “specific” means the measure is a bulk property (an intensive property), wherein the quantity of substance must be specified. For example, the heat energy required to raise water’s temperature one kelvin (equal to one Celsius degree) is 4184 joules per kilogram—the kilogram being the specified quantity. Scientifically, this measure would be expressed as c = 4184 J kg–1 K–1.
Basic metrics of specific heat capacity
Unit quantity
Because there is disagreement as to whether specific heat capacity is defined with regard to mass, number of atoms, or number of molecules, some people use the term "unit quantity" as a generic term for all possible physical quantities. When measuring specific heat capacity in science and engineering, the unit quantity of a substance is often in terms of mass: either the gram or kilogram, both of which are an SI unit. Especially in chemistry though, the unit quantity of specific heat capacity may also be the mole, which is a certain number of molecules or atoms. When the unit quantity is the mole, the term molar heat capacity may also be used to more explicitly describe the measure.
Temperature interval
The temperature interval in science, engineering and chemistry is usually one kelvin or degree Celsius (both of which have the same magnitude).
Other units
In the U.S., other units of measure for specific heat capacity are typically used in disciplines such as construction and civil engineering. There, the mass quantity is often the pound-mass, the unit of heat energy is the British thermal unit, and the temperature interval is the degree Fahrenheit.
If temperature is expressed in natural rather than historical terms i.e. as a rate of energy increase per unit increase in state uncertainty, then heat capacity becomes the number of bits of mutual information between system and surroundings lost per two-fold increase in absolute temperature[2]. Thus for instance, with each 2-fold increase in absolute temperature we lose 3/2 bits of mutual information per atom in a monatomic ideal gas.
Basic equations
- The equation relating heat energy to specific heat capacity, where the unit quantity is in terms of mass is:
-
- where
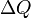 is the heat energy put into or taken out of the substance,
is the heat energy put into or taken out of the substance, 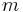 is the mass of the substance,
is the mass of the substance, 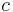 is the specific heat capacity, and
is the specific heat capacity, and 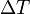 is the temperature differential where the initial temperature of the reaction is subtracted from the final temperature.
is the temperature differential where the initial temperature of the reaction is subtracted from the final temperature.
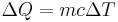
- Where the unit quantity is in terms of moles, the equation relating heat energy to specific heat capacity (also known as molar heat capacity) is
-
- where is the heat energy put into or taken out of the substance,
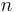 is the number of moles, is the specific heat capacity, and is the temperature differential.
is the number of moles, is the specific heat capacity, and is the temperature differential.
Factors that affect specific heat capacity

- Degrees of freedom: Molecules are quite different from the monatomic gases like helium and argon. With monatomic gases, heat energy comprises only translational motions. Translational motions are ordinary, whole-body movements in 3D space whereby particles move about and exchange energy in collisions—like rubber balls in a vigorously shaken container (see animation here). These simple movements in the three X, Y, and Z–axis dimensions of space means monatomic atoms have three translational degrees of freedom. Molecules, however, have various internal vibrational and rotational degrees of freedom because they are complex objects; they are a population of atoms that can move about within a molecule in different ways (see animation at right). Heat energy is stored in these internal motions, but on a per-atom basis, the heat capacity of molecules does not exceed the heat capacity of monatomic gases, unless vibrational modes are brought into play.
For instance, nitrogen, which is a diatomic molecule, has five active degrees of freedom at room temperature: the three comprising translational motion plus two rotational degrees of freedom internally. Although the constant-volume molar heat capacity of nitrogen at this temperature is five-thirds that of monatomic gases, on a per-mole of atoms basis, it is only five-sixths that of a monatomic gas, because each atom has fewer degrees of freedom at low temperatures, due to the intra-atomic bond. This is expected since two nitrogen atoms would have six degrees of freedom (three for each atom) if free, but when locked together, they lose one degree of freedom, and together possess only five.
At higher temperatures, however, nitrogen gas gains two more degrees of internal freedom, as the molecule is excited into higher vibrational modes which store heat energy, and then the heat capacity per volume or mole of molecules approaches seven-thirds that of monatomic gases, or seven-sixths of monatomic, on a mole-of-atoms basis. This is now a higher heat capacity per atom than the monatomic figure, because the vibrational mode enables an extra degree of potential energy freedom per pair of atoms, which monatomic gases cannot possess. [3] See Thermodynamic temperature for more information on translational motions, kinetic (heat) energy, and their relationship to temperature.
- Per mole of molecules: When the specific heat capacity, c, of a material is measured (lowercase c means the unit quantity is in terms of mass), different values arise because different substances have different molar masses (essentially, the weight of the individual atoms or molecules). Heat energy arises, in part, due to the number of atoms or molecules that are vibrating. If a substance has a lighter molar mass, then each gram of it has more atoms or molecules available to store heat energy. This is why hydrogen—the lightest substance there is—has such a high specific heat capacity on a gram basis; one gram of it contains twice the number of molecules as a gram of helium, and the ratio for other substances is even larger. If specific heat capacity is measured on a molar basis (uppercase C), however, the differences between substances is less pronounced and hydrogen’s molar heat capacity is unremarkable.
- Per mole of atoms: Conversely, for molecular-based substances (which also absorb heat into their internal degrees of freedom), massive, complex molecules with high atomic count—like gasoline—can store a great deal of energy per mole and yet are quite unremarkable on a mass basis, or on a per-atom basis. This is because, in fully excited systems, heat is stored independently by each atom in a substance, not primarily by the bulk motion of molecules.
Thus, it is the heat capacity per-mole-of-atoms, not per-mole-of-molecules, which comes closest to being a constant for all substances at high temperatures. For this reason, some care should be taken to specify a mole-of-molecules basis vs. a mole-of-atoms basis, when comparing specific heat capacities of molecular solids and gases. Ideal gases have the same numbers of molecules per volume, so increasing molecular complexity adds heat capacity on a per-volume and per-mole-of-molecules basis, but may lower or raise heat capacity on a per-atom basis, depending on whether the temperature is sufficient to store energy as atomic vibration.
In solids, the limit of heat capacity in general is about 3R per mole of atoms, as 6 degrees of freedom (3 kinetic and 3 potential) are available to each atom, and each of these contributes R/2 specific heat capacity per mole of atoms. For monatomic gases, the specific heat is only half of this (3R/2 per mole) due to loss of all potential energy degrees of freedom. For polyatomic gases, the heat capacity will be intermediate between these values on a per-mole-of-atoms basis, and (for heat-stable molecules) would approach the limit of 3R per mole of atoms for gases composed of complex molecules with all vibrational modes excited. This is because complex gas molecules may be thought of as large blocks of matter which have lost only a small fraction of degrees of freedom, as compared to the fully integrated solid.
Corrolaries of these considerations, for solids: Since the bulk density of a solid chemical element is strongly related to its molar mass, generally speaking, there is a strong, inverse correlation between a solid’s density and its cp (constant-pressure specific heat capacity on a mass basis). Large ingots of low-density solids tend to absorb more heat energy than a small, dense ingot of the same mass because the former usually has more atoms. Thus, generally speaking, there a close correlation between the size of a solid chemical element and its total heat capacity (see Volumetric heat capacity). There are however, many departures from the general trend. For instance, arsenic, which is only 14.5% less dense than antimony, has nearly 59% more specific heat capacity on a mass basis. In other words; even though an ingot of arsenic is only about 17% larger than an antimony one of the same mass, it absorbs about 59% more heat energy for a given temperature rise.
Other factors
- Hydrogen bonds: Hydrogen-containing polar molecules like ethanol, ammonia, and water have powerful, intermolecular hydrogen bonds when in their liquid phase. These bonds provide another place where heat energy may be stored as potential energy of vibration, even at comparatively low temperatures. Hydrogen bonds account for the fact that liquid water stores nearly the theoretical limit of 3R per mole of atoms, even at relatively low temperatures (i.e. near the freezing point of water).
- Impurities: In the case of alloys, there are several conditions in which small impurity concentrations can greatly affect the specific heat. Alloys may exhibit marked difference in behaviour even in the case of small amounts of impurities being one element of the alloy; for example impurities in semiconducting ferromagnetic alloys may lead to quite different specific heat properties as first predicted by White and Hogan.[4]
Symbols and standards
When mass is the unit quantity, the symbol for specific heat capacity is lowercase c. When the mole is the unit quantity, the symbol is uppercase C. Alternatively—especially in chemistry as opposed to engineering—the uppercase version for specific heat, C, may be used in combination with a suffix representing enthalpy (symbol: either H or h); specifically, when the mole is the unit quantity, the enthalpy suffix is uppercase H and when mass is the unit quantity, the suffix is lowercase h.
The modern SI units for measuring specific heat capacity are either the joule per gram-kelvin (J g–1 K–1) or the joule per mole-kelvin (J mol–1 K–1). The various SI prefixes can create variations of these units (such as kJ kg–1 K–1 and kJ mol–1 K–1). Symbols for alternative units are as follows: pounds-mass (symbol: lb) for quantity, calories (symbol: cal) and British thermal units (symbol: BTU) for energy, and degree Fahrenheit (symbol: °F) for the increment of temperature.
There are two distinctly different experimental conditions under which specific heat capacity is measured and these are denoted with a subscripted suffix modifying the symbols C or c. The specific heat of substances are typically measured under constant pressure (Symbols: Cp or cp). However, fluids (gases and liquids) are typically also measured at constant volume (Symbols: Cv or cv). Measurements under constant pressure produces greater values than those at constant volume because work must be performed in the former. This difference is particularly great in gases where values under constant pressure are typically 30% to 66.7% greater than those at constant volume.
Thus, the symbols for specific heat capacity are as follows:
| Under constant pressure |
At constant volume |
|
| Unit quantity = mole | Cp or CpH | Cv or CvH |
| Unit quantity = mass | cp or Cph | cv or Cvh |
The ratio of the specific heats (or Heat capacity ratio) is usually denoted by 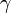 (gamma). It is often used in equations, such as for calculating speed of sound in an ideal gas.
(gamma). It is often used in equations, such as for calculating speed of sound in an ideal gas.
The specific heat capacities of substances comprising molecules (distinct from the monatomic gases) are not fixed constants and vary somewhat depending on temperature. Accordingly, the temperature at which the measurement is made is usually also specified. Examples of two common ways to cite the specific heat of a substance are as follows:
Water (liquid): cp = 4.1855 J g–1 K–1 (15 °C), and…
Water (liquid): CvH = 74.539 J mol–1 K–1 (25 °C)
The pressure at which specific heat capacity is measured is especially important for gases and liquids. The standard pressure was once virtually always “one standard atmosphere” which is defined as the sea level–equivalent value of precisely 101.325 kPa (760 Torr). In the case of water, 101.325 kPa is still typically used due to water’s unique role in temperature and physical standards. However, in 1982, the International Union of Pure and Applied Chemistry (IUPAC) recommended that for the purposes of specifying the physical properties of substances, “the standard pressure” should be defined as precisely 100 kPa (≈750.062 Torr).[5] Besides being a round number, this had a very practical effect: relatively few people live and work at precisely sea level; 100 kPa equates to the mean pressure at an altitude of about 112 metres (which is closer to the 194–metre, world–wide median altitude of human habitation). Accordingly, the pressure at which specific heat capacity is measured should be specified since one can not assume its value. An example of how pressure is specified is as follows:
Water (gas): CvH = 28.03 J mol–1 K–1 (100 °C, 101.325 kPa)
Note in the above specification that the experimental condition is at constant volume. Still, the pressure within this fixed volume is controlled and specified.
Heat capacity
Heat capacity (symbol: Cp) — as distinct from specific heat capacity — is the measure of the heat energy required to increase the temperature of an object by a certain temperature interval. Heat capacity is an extensive property because its value is proportional to the amount of material in the object; for example, a bathtub of water has a greater heat capacity than a cup of water.
Heat capacity is usually expressed in units of J K–1 (or J/K), subject to the caveats and exceptions detailed in both Basic metrics of specific heat capacity and Symbols and standards, above. For instance, one could write that the gasoline in a 55-gallon drum has an average heat capacity of 347 kJ/K.
Physical properties cannot be measured with 100% accuracy. Accordingly, it is usually unnecessary as a practical matter, to specify the defined state at which the measurement was made; e.g. “(25 °C, 100 kPa).” In most cases, it is assumed that the substance’s specific heat capacity is a published value and the object’s quantity is subject to such a sizable relative uncertainty that it renders this detail moot. An exception would be when an object has an accurately known or precisely defined quantity; e.g. “The heat capacity of the International Prototype Kilogram is 133 J/K (25 °C).” Another exception would be when the defined state varies significantly from standard conditions.
Table of specific heat capacities
Note that especially high values, as for paraffin, water and ammonia, result from calculating specific heats in terms of moles of molecules. If specific heat is expressed per mole of atoms for these substances, few values exceed the theoretical Dulong-Petit limit of 25 J/K/mole = 3 R per mole.
| Substance | Phase | Cp kJ kg−1 K−1 |
Cp,m J mol−1 K−1 |
Cv,m J mol−1 K−1 |
Volumetric heat capacity J cm−3 K−1 |
|---|---|---|---|---|---|
| Air (Sea level, dry, 0 °C) | gas | 1.0035 | 29.07 | 20.7643 | 0.001297 |
| Air (typical room conditionsA) | gas | 1.012 | 29.19 | 20.85 | |
| Aluminium | solid | 0.897 | 24.2 | 2.422 | |
| Ammonia | liquid | 4.700 | 80.08 | 3.263 | |
| Antimony | solid | 0.207 | 25.2 | 1.386 | |
| Argon | gas | 0.5203 | 20.7862 | 12.4717 | |
| Arsenic | solid | 0.328 | 24.6 | 1.878 | |
| Beryllium | solid | 1.82 | 16.4 | 3.367 | |
| Bismuth[6] | solid | 0.123 | 25.7 | 1.20 | |
| Copper | solid | 0.385 | 24.47 | 3.45 | |
| Carbon dioxide CO2[7] | gas | 0.839* | 36.94 | 28.46 | |
| Diamond | solid | 0.5091 | 6.115 | 1.782 | |
| Ethanol | liquid | 2.44 | 112 | 1.925 | |
| Gasoline | liquid | 2.22 | 228 | 1.64 | |
| Glass[6] | solid | 0.84 | |||
| Gold | solid | 0.1291 | 25.42 | 2.492 | |
| Granite[6] | solid | 0.790 | 2.17 | ||
| Graphite | solid | 0.710 | 8.53 | 1.534 | |
| Helium | gas | 5.1932 | 20.7862 | 12.4717 | |
| Hydrogen | gas | 14.30 | 28.82 | ||
| Hydrogen sulfide H2S[7] | gas | 1.015* | 34.60 | ||
| Iron | solid | 0.450 | 25.1 | 3.537 | |
| Lead | solid | 0.127 | 26.4 | 1.44 | |
| Lithium | solid | 3.58 | 24.8 | 1.912 | |
| Magnesium | solid | 1.02 | 24.9 | 1.773 | |
| Mercury | liquid | 0.1395 | 27.98 | 1.888 | |
| Nitrogen | gas | 1.040 | 29.12 | 20.8 | |
| Neon | gas | 1.0301 | 20.7862 | 12.4717 | |
| Oxygen | gas | 0.918 | 29.38 | ||
| Paraffin wax | solid | 2.5 | 900 | 2.325 | |
| Silica (fused) | solid | 0.703 | 42.2 | 1.547 | |
| Silver[6] | solid | 0.233 | 24.9 | 2.44 | |
| Tungsten[6] | solid | 0.134 | 24.8 | 2.58 | |
| Uranium | solid | 0.116 | 27.7 | 2.216 | |
| Water (steam) | gas (100 °C) | 2.080 | 37.47 | 28.03 | |
| Water | liquid (25 °C) | 4.1813 | 75.327 | 74.53 | 4.186 |
| Water (ice)[6] | solid (-10 °C) | 2.050 | 38.09 | 1.938 | |
| Zinc[6] | solid | 0.387 | 25.2 | 2.76 | |
| All measurements are at 25 °C unless otherwise noted. Notable minima and maxima are shown in maroon. |
|||||
A Assuming an altitude of 194 metres above mean sea level (the world–wide median altitude of human habitation), an indoor temperature of 23 °C, a dewpoint of 9 °C (40.85% relative humidity), and 760 mm–Hg sea level–corrected barometric pressure (molar water vapor content = 1.16%).
*Derived data by calculation
Specific heat capacity of building materials
(Usually of interest to builders and solar designers)
| Substance | Phase | cp J g−1 K−1 |
|---|---|---|
| Asphalt | solid | 0.92 |
| Brick | solid | 0.84 |
| Concrete | solid | 0.88 |
| Glass, silica | solid | 0.84 |
| Glass, crown | solid | 0.67 |
| Glass, flint | solid | 0.503 |
| Glass, pyrex | solid | 0.753 |
| Granite | solid | 0.790 |
| Gypsum | solid | 1.09 |
| Marble, mica | solid | 0.880 |
| Sand | solid | 0.835 |
| Soil | solid | 0.80 |
| Wood | solid | 0.42 |
Derivations of heat capacity and specific heat capacity
Definition of heat capacity
Heat capacity is mathematically defined as the ratio of a small amount of heat δQ added to the body, to the corresponding small increase in its temperature dT:
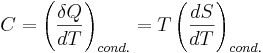
For thermodynamic systems with more than one physical dimension, the above definition does not give a single, unique quantity unless a particular infinitesimal path through the system’s phase space has been defined (this means that one needs to know at all times where all parts of the system are, how much mass they have, and how fast they are moving). This information is used to account for different ways that heat can be stored as kinetic energy (energy of motion) and potential energy (energy stored in force fields), as an object expands or contracts. For all real systems, the path through these changes must be explicitly defined, since the value of heat capacity depends on which path from one temperature to another, is chosen. Of particular usefulness in this context are the values of heat capacity for constant volume, CV, and constant pressure, CP. These will be defined below.
Heat capacity of compressible bodies
The state of a simple compressible body with fixed mass is described by two thermodynamic parameters such as temperature T and pressure p. Therefore as mentioned above, one may distinguish between heat capacity at constant volume, 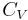 , and heat capacity at constant pressure,
, and heat capacity at constant pressure, 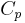 :
:
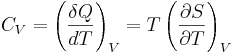
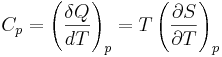
where
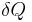 is the infinitesimal amount of heat added,
is the infinitesimal amount of heat added,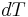 is the subsequent rise in temperature.
is the subsequent rise in temperature.
The increment of internal energy is the heat added and the work added:
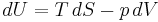
So the heat capacity at constant volume is
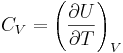
The enthalpy is defined by 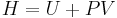 . The increment of enthalpy is
. The increment of enthalpy is
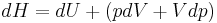
which, after replacing dU with the equation above and cancelling the PdV terms reduces to:
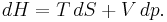
So the heat capacity at constant pressure is
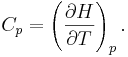
Note that this last “definition” is a bit circular, since the concept of “enthalpy” itself was invented to be a measure of heat absorbed or produced at constant pressures (the conditions in which chemists usually work). As such, enthalpy merely accounts for the extra heat which is produced or absorbed by pressure-volume work at constant pressure. Thus, it is not surprising that constant-pressure heat capacities may be defined in terms of enthalpy, since “enthalpy” was defined in the first place to make this so.
Relation between specific heats
Measuring the heat capacity at constant volume can be prohibitively difficult for liquids and solids. That is, small temperature changes typically require large pressures to maintain a liquid or solid at constant volume implying the containing vessel must be nearly rigid or at least very strong (see coefficient of thermal expansion and compressibility). Instead it is easier to measure the heat capacity at constant pressure (allowing the material to expand or contract as it wishes) and solve for the heat capacity at constant volume using mathematical relationships derived from the basic thermodynamic laws. Starting from the fundamental Thermodynamic Relation one can show,
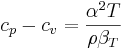
where,
 is the coefficient of thermal expansion, and
is the coefficient of thermal expansion, and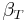 is the isothermal compressibility.
is the isothermal compressibility.
A derivation is given here For an ideal gas this reduces to the simple relation,
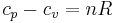 , where n is the number of moles, and
, where n is the number of moles, and 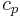 and
and 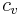 are extensive (non-molar) properties.
are extensive (non-molar) properties.
Specific heat capacity
The specific heat capacity of a material is
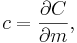
which in the absence of phase transitions is equivalent to
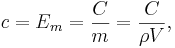
where
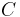 is the heat capacity of a body made of the material in question,
is the heat capacity of a body made of the material in question,- is the mass of the body,
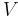 is the volume of the body, and
is the volume of the body, and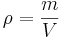 is the density of the material.
is the density of the material.
For gases, and also for other materials under high pressures, there is need to distinguish between different boundary conditions for the processes under consideration (since values differ significantly between different conditions). Typical processes for which a heat capacity may be defined include isobaric (constant pressure, 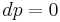 ) or isochoric (constant volume,
) or isochoric (constant volume, 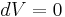 ) processes. The corresponding specific heat capacities are expressed as
) processes. The corresponding specific heat capacities are expressed as
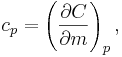
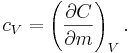
A related parameter to is 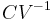 , the volumetric heat capacity. In engineering practice,
, the volumetric heat capacity. In engineering practice, 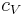 for solids or liquids often signifies a volumetric heat capacity, rather than a constant-volume one. In such cases, the mass-specific heat capacity (specific heat) is often explicitly written with the subscript , as
for solids or liquids often signifies a volumetric heat capacity, rather than a constant-volume one. In such cases, the mass-specific heat capacity (specific heat) is often explicitly written with the subscript , as 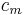 . Of course, from the above relationships, for solids one writes
. Of course, from the above relationships, for solids one writes
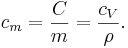
Dimensionless heat capacity
The dimensionless heat capacity of a material is
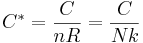
where
- C is the heat capacity of a body made of the material in question (J·K−1)
- n is the amount of matter in the body (mol)
- R is the gas constant (J·K−1·mol−1)
- nR=Nk is the amount of matter in the body (J·K−1)
- N is the number of molecules in the body. (dimensionless)
- k is Boltzmann’s constant (J·K−1·molecule−1)
Again, SI units shown for example.
Theoretical models
Gas phase
The specific heat of the gas is best conceptualized in terms of the degrees of freedom of an individual molecule. The different degrees of freedom correspond to the different ways in which the molecule may store energy. The molecule may store energy in its translational motion according to the formula:
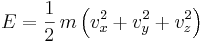
where m is the mass of the molecule and ![[v_x,v_y,v_z]](/2009-wikipedia_en_wp1-0.7_2009-05/I/7a82c338b51bd7c2b044421148b81c62.png) is velocity of the center of mass of the molecule. Each direction of motion constitutes a degree of freedom, so that there are three translational degrees of freedom.
is velocity of the center of mass of the molecule. Each direction of motion constitutes a degree of freedom, so that there are three translational degrees of freedom.
In addition, a molecule may have rotational motion. The kinetic energy of rotational motion is generally expressed as
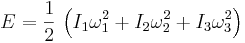
where I is the moment of inertia tensor of the molecule, and ![[\omega_1,\omega_2,\omega_3]](/2009-wikipedia_en_wp1-0.7_2009-05/I/f4b1f1bb48659c4ea73967b5f7cc792b.png) is the angular velocity pseudo-vector (in a coordinate system aligned with the principle axes of the molecule). In general, then, there will be three additional degrees of freedom corresponding to the rotational motion of the molecule, (For linear molecules one of the inertia tensor terms vanishes and there are only two rotational degrees of freedom). The degrees of freedom corresponding to translations and rotations are called the “rigid” degrees of freedom, since they do not involve any deformation of the molecule.
is the angular velocity pseudo-vector (in a coordinate system aligned with the principle axes of the molecule). In general, then, there will be three additional degrees of freedom corresponding to the rotational motion of the molecule, (For linear molecules one of the inertia tensor terms vanishes and there are only two rotational degrees of freedom). The degrees of freedom corresponding to translations and rotations are called the “rigid” degrees of freedom, since they do not involve any deformation of the molecule.
The motions of the atoms in a molecule which are not part of its gross translational motion or rotation may be classified as vibrational motions. It can be shown that if there are n atoms in the molecule, there will be as many as 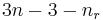 vibrational degrees of freedom, where
vibrational degrees of freedom, where 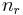 is the number of rotational degrees of freedom. The actual number may be less due to various symmetries.
is the number of rotational degrees of freedom. The actual number may be less due to various symmetries.
If the molecule could be entirely described using classical mechanics, then we could use the theorem of equipartition of energy to predict that each degree of freedom would have an average energy in the amount of (1/2)kT where k is Boltzmann’s constant and T is the temperature. Our calculation of the heat content would be straightforward. Each molecule would be holding, on average, an energy of (f/2)kT where f is the total number of degrees of freedom in the molecule. The total internal energy of the gas would be (f/2)NkT where N is the total number of molecules. The heat capacity (at constant volume) would then be a constant (f/2)Nk , the specific heat capacity would be (f/2)k and the dimensionless heat capacity would be just f/2.
The various degrees of freedom cannot generally be considered to obey classical mechanics. Classically, the energy residing in each degree of freedom is assumed to be continuous - it can take on any positive value, depending on the temperature. In reality, the amount of energy that may reside in a particular degree of freedom is quantized: It may only be increased and decreased in finite amounts. A good estimate of the size of this minimum amount is the energy of the first excited state of that degree of freedom above its ground state. For example, the first vibrational state of the hydrogen chloride (HCl) molecule has an energy of about 5.74 × 10–20 joule. If this amount of energy were deposited in a classical degree of freedom, it would correspond to a temperature of about 4156 K.
If the temperature of the substance is so low that the equipartition energy of (1/2)kT is much smaller than this excitation energy, then there will be little or no energy in this degree of freedom. This degree of freedom is then said to be “frozen out". As mentioned above, the temperature corresponding to the first excited vibrational state of HCl is about 4156 K. For temperatures well below this value, the vibrational degrees of freedom of the HCl molecule will be frozen out. They will contain little energy and will not contribute to the heat content of the HCl gas.
It can be seen that for each degree of freedom there is a critical temperature at which the degree of freedom “unfreezes” and begins to accept energy in a classical way. In the case of translational degrees of freedom, this temperature is that temperature at which the thermal wavelength of the molecules is roughly equal to the size of the container. For a container of macroscopic size (e.g. 10 cm) this temperature is extremely small and has no significance, since the gas will certainly liquify or freeze before this low temperature is reached. For any real gas we may consider translational degrees of freedom to always be classical and contain an average energy of (3/2)kT per molecule.
The rotational degrees of freedom are the next to “unfreeze". In a diatomic gas, for example, the critical temperature for this transition is usually a few tens of kelvins, although with a very light molecule such as hydrogen the rotational energy levels will be spaced so widely that rotational heat capacity may not completely "unfreeze" until considerably higher temperatures are reached. Finally, the vibrational degrees of freedom are generally the last to unfreeze. As an example, for diatomic gases, the critical temperature for the vibrational motion is usually a few thousands of kelvins, although with unusually heavy gases such as iodine gas I2, or bromine gas Br2, some vibrational heat capacity may be observed even at room temperatures.
It should be noted that it has been assumed that atoms have no rotational or internal degrees of freedom. This is in fact untrue. For example, atomic electrons can exist in excited states and even the atomic nucleus can have excited states as well. Each of these internal degrees of freedom are assumed to be frozen out due to their relatively high excitation energy. Nevertheless, for sufficiently high temperatures, these degrees of freedom cannot be ignored. In a few exceptional cases, such molecular transitions are of sufficiently such low energy that they contribute to heat capacity at room temperature, or even at cryogenic temperatures.
One example of an electronic transition which contributes heat capacity at standard temperature is that of nitric oxide (NO), in which the single electron in an anti-bonding molecular orbital has energy transitions which contribute to the heat capacity of the gas even at room temperature.
An example of a nuclear magnetic transition which is of importance is the transition which converts the spin isomers of hydrogen gas to each other. At room temperature, the proton spins of hydrogen gas are alligned 75% of the time, resulting in orthohydrogen. However, at liquid hydrogen temperatures, the parahydrogen form of H2 in which spins are anti-alligned predominates, and the heat capacity of the transistion is sufficient to boil the hydrogen if this is heat is not removed with a catalyst, after the gas has been condensed. This example also illustrates the fact that some modes of storage of heat may not be in constant equilibrium with each other in substances, and heat absorbed or released from such phase changes may "catch up" with temperature changes of substances only after a certain time.
Monatomic gas
In the case of a monatomic gas such as helium under constant volume, if it assumed that no electronic or nuclear quantum excitations occur, each atom in the gas has only 3 degrees of freedom, all of a translational type. No energy dependence is associated with the degrees of freedom which define the position of the atoms. While, in fact, the degrees of freedom corresponding to the momenta of the atoms are quadratic, and thus contribute to the heat capacity. There are N atoms, each of which has 3 components of momentum, which leads to 3N total degrees of freedom. This gives:
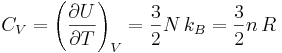
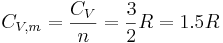
where
- is the heat capacity at constant volume of the gas
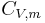 is the molar heat capacity at constant volume of the gas
is the molar heat capacity at constant volume of the gas- N is the total number of atoms present in the container
- n is the number of moles of atoms present in the container (n is the ratio of N and Avogadro’s number)
- R is the ideal gas constant, (8.314570[70] J K−1mol−1). R is equal to the product of Boltzmann’s constant
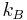 and Avogadro’s number
and Avogadro’s number
The following table shows experimental molar constant volume heat capacity measurements taken for each noble monatomic gas (at 1 atm and 25 °C):
| Monatomic gas | CV, m (J K−1 mol−1) | CV, m/R |
|---|---|---|
| He | 12.5 | 1.50 |
| Ne | 12.5 | 1.50 |
| Ar | 12.5 | 1.50 |
| Kr | 12.5 | 1.50 |
| Xe | 12.5 | 1.50 |
It is apparent from the table that the experimental heat capacities of the monatomic noble gases agrees with this simple application of statistical mechanics to a very high degree.
Diatomic gas
In the somewhat more complex case of an ideal gas of diatomic molecules, the presence of internal degrees of freedom are apparent. In addition to the three translational degrees of freedom, there are rotational and vibrational degrees of freedom. In general, the number of degrees of freedom, f, in a molecule with na atoms is 3na:
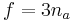
Mathematically, there are a total of three rotational degrees of freedom, one corresponding to rotation about each of the axes of three dimensional space. However, in practice we shall only consider the existence of two degrees of rotational freedom for linear molecules. This approximation is valid because the moment of inertia about the internuclear axis is vanishingly small with respect other moments of inertia in the molecule (this is due to the extremely small radii of the atomic nuclei, compared to the distance between them in a molecule). Quantum mechanically, it can be shown that the interval between successive rotational energy eigenstates is inversely proportional to the moment of inertia about that axis. Because the moment of inertia about the internuclear axis is vanishingly small relative to the other two rotational axes, the energy spacing can be considered so high that no excitations of the rotational state can possibly occur unless the temperature is extremely high. We can easily calculate the expected number of vibrational degrees of freedom (or vibrational modes). There are three degrees of translational freedom, and two degrees of rotational freedom, therefore
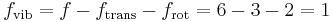
Each rotational and translational degree of freedom will contribute R/2 in the total molar heat capacity of the gas. Each vibrational mode will contribute  to the total molar heat capacity, however. This is because for each vibrational mode, there is a potential and kinetic energy component. Both the potential and kinetic components will contribute R/2 to the total molar heat capacity of the gas. Therefore, we expect that a diatomic molecule would have a molar constant-volume heat capacity of
to the total molar heat capacity, however. This is because for each vibrational mode, there is a potential and kinetic energy component. Both the potential and kinetic components will contribute R/2 to the total molar heat capacity of the gas. Therefore, we expect that a diatomic molecule would have a molar constant-volume heat capacity of
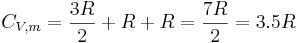
where the terms originate from the translational, rotational, and vibrational degrees of freedom, respectively.
The following is a table of some molar constant-volume heat capacities of various diatomic gasses
| Diatomic gas | CV, m (J K−1 mol−1) | CV, m / R |
|---|---|---|
| H2 | 20.18 | 2.427 |
| CO | 20.2 | 2.43 |
| N2 | 19.9 | 2.39 |
| Cl2 | 24.1 | 2.90 |
| Br2 | 32.0 | 3.84 |
From the above table, clearly there is a problem with the above theory. All of the diatomics examined have heat capacities that are lower than those predicted by the Equipartition Theorem, except Br2. However, as the atoms composing the molecules become heavier, the heat capacities move closer to their expected values. One of the reasons for this phenomenon is the quantization of vibrational, and to a lesser extent, rotational states. In fact, if it is assumed that the molecules remain in their lowest energy vibrational state because the inter-level energy spacings are large, the predicted molar constant volume heat capacity for a diatomic molecule becomes
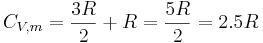
which is a fairly close approximation of the heat capacities of the lighter molecules in the above table. If the quantum harmonic oscillator approximation is made, it turns out that the quantum vibrational energy level spacings are actually inversely proportional to the square root of the reduced mass of the atoms composing the diatomic molecule. Therefore, in the case of the heavier diatomic molecules, the quantum vibrational energy level spacings become finer, which allows more excitations into higher vibrational levels at a fixed temperature.
Other gases
In summary, the heat capacity of an ideal gas with f degrees of freedom is given by
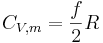
This equation also applies to polyatomic gases, if the degrees of freedom are known.[7]
Solid phase
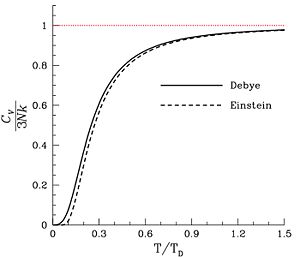
For matter in a crystalline solid phase, the Dulong-Petit law, which was discovered empirically, states that the mole-specific heat capacity assumes the value 3 R. Indeed, for solid metallic chemical elements at room temperature, molar heat capacities range from about 2.8 R to 3.4 R (beryllium being a notable exception at 2.0 R).
The theoretical maximum heat capacity for larger and larger multi-atomic gases at higher temperatures, also approaches the Dulong-Petit limit of 3 R, so long as this is calculated per mole of atoms, not molecules. The reason is that gases with very large molecules, in theory have almost the same high-temperature heat capacity as solids, lacking only the (small) heat capacity contribution that comes from potential energy that cannot be stored between separate molecules in a gas.
The Dulong-Petit “limit” results from the equipartition theorem, and as such is only valid in the classical limit of a microstate continuum, which is a high temperature limit. For light and non-metallic elements, as well as most of the common molecular solids based on carbon compounds at standard ambient temperature, quantum effects may also play an important role, as they do in multi-atomic gases. These effects usually combine to give heat capacities lower than 3 R per mole of atoms in the solid, although heat capacities calculated per mole of molecules in molecular solids may be more than 3 R. For example, the heat capacity of water ice at the melting point is about 4.6 R per mole of molecules, but only 1.5 R per mole of atoms. The lower number results from the “freezing out” of possible vibration modes for light atoms at suitably low temperatures, just as in many gases. These effects are seen in solids more often than liquids: for example the heat capacity of liquid water is again close to the theoretical 3 R per mole of atoms of the Dulong-Petit theoretical maximum.
For a more modern and precise analysis of the heat capacities of solids, especially at low temperatures, it is useful to use the idea of phonons. See Debye model.
Heat capacity at absolute zero
From the definition of entropy
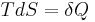
we can calculate the absolute entropy by integrating from zero temperature to the final temperature Tf
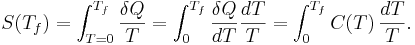
{kind=link}
The heat capacity must be zero at zero temperature in order for the above integral not to yield an infinite absolute entropy, thus violating the third law of thermodynamics. One of the strengths of the Debye model is that (unlike the preceding Einstein model) it predicts an approach of heat capacity toward zero as zero temperature is approached, and also predicts the proper mathematical form of this approach.
See also
|
|
References
- ↑ Laider, Keith, J. (1993). The World of Physical Chemistry. Oxford University Press. ISBN 0-19-855919-4.
- ↑ P. Fraundorf (2003) Heat capacity in bits, American Journal of Physics 71:11, 1142-1151. (arXiv:cond-mat/9711074)
- ↑ The comparison must be made under constant-volume conditions — CvH — so that no work is performed. Nitrogen’s CvH (100 kPa, 20 °C) = 20.8 J mol–1 K–1 vs. the monatomic gases which equal 12.4717 J mol–1 K–1. Citations: W.H. Freeman’s Physical Chemistry, Part 3: Change (422 kB PDF, here), Exercise 21.20b, Pg. 787. Also Georgia State University’s Molar Specific Heats of Gases.
- ↑ C. Michael Hogan, (1969) Density of States of an Insulating Ferromagnetic Alloy Phys. Rev. 188, 870 - 874, [Issue 2 – December 1969
- ↑ IUPAC.org, Gold Book, Standard Pressure
- ↑ 6.0 6.1 6.2 6.3 6.4 6.5 6.6 Table of Specific Heats
- ↑ 7.0 7.1 7.2 Textbook: Young and Geller College Physics, 8e, Pearson Education, 2008