Sickle-cell disease
| Sickle-cell disease Classification and external resources |
||
 |
||
|---|---|---|
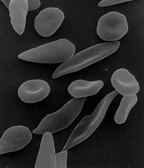
| Sickle-shaped red blood cells | ||
| ICD-10 | D57. | |
| ICD-9 | 282.6 | |
| OMIM | 603903 | |
| DiseasesDB | 12069 | |
| MedlinePlus | 000527 | |
| eMedicine | med/2126 oph/490 ped/2096 emerg/26 emerg/406 | |
| MeSH | C15.378.071.141.150.150 | |
Sickle-cell disease or sickle-cell anaemia (or anemia) is a blood disorder characterized by red blood cells that assume an abnormal, rigid, sickle shape. Sickling decreases the cells' flexibility and results in a risk of various other complications. Life expectancy is shortened, with older studies reporting an average life expectancy of 42 and 48 years for males and females, respectively.[1]
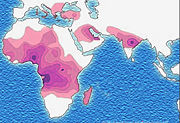
Sickle-cell disease occurs more commonly in people (or their descendants) from parts of sub-Saharan Africa, where malaria is or was common, but it also occurs in people of other ethnicities. This is because those with one or two alleles of the sickle-cell disease are resistant to malaria since the sickle red blood cells are not conducive to the parasites - in areas where malaria is common, there is a survival value in carrying a single sickle-cell gene, AS heterozygotes.
Contents |
Classification
Sickle-cell anaemia is the name of a specific form of sickle-cell disease in which there is homozygosity for the mutation that causes HbS. Sickle-cell anaemia is also referred to as "HbSS", "SS disease", "haemoglobin S", or permutations thereof. Other, rarer forms of sickle-cell disease include sickle-haemoglobin C disease (HbSC), sickle beta-plus-thalassaemia (HbS/β+) and sickle beta-zero-thalassaemia (HbS/β0). These other forms of sickle-cell disease are compound heterozygous states in which the person has only one copy of the mutation that causes HbS and one copy of another abnormal haemoglobin allele.
The term "disease" is applied since the inherited abnormality causes a pathological condition that can lead to death and severe complications. Not all inherited variants of haemoglobin are detrimental, a concept known as genetic polymorphism.
Signs and symptoms
Sickle-cell disease may lead to various acute and chronic complications, several of which are potentially lethal.
Vaso-occlusive crisis
The vaso-occlusive crisis is caused by sickle-shaped red blood cells that obstruct capillaries and restrict blood flow to an organ, resulting in ischemia, pain, and organ damage. The frequency, severity, and duration of these crises vary considerably. Painful crises are treated with hydration and analgesics; pain management requires opioid administration at regular intervals until the crisis has settled. For milder crises a subgroup of patients manage on NSAIDs (such as diclofenac or naproxen). For more severe crises most patients require inpatient management for intravenous opioids; patient-controlled analgesia (PCA) devices are commonly used in this setting. Diphenhydramine is sometimes effective for the itching associated with the opioid use. Incentive spirometry, a technique to encourage deep breathing to minimise the development of atelectasis, is recommended.
Because of its narrow vessels and function in clearing defective red blood cells, the spleen is frequently affected. It is usually infarcted before the end of childhood in individuals suffering from sickle-cell anaemia. This autosplenectomy increases the risk of infection from encapsulated organisms;[2][3] preventive antibiotics and vaccinations are recommended for those with such asplenia.
A recognised type of sickle crisis is the acute chest syndrome, a condition characterised by fever, chest pain, difficulty breathing, and pulmonary infiltrate on a chest X-ray. Given that pneumonia and sickling in the lung can both produce these symptoms, the patient is treated for both conditions. It can be triggered by painful crisis, respiratory infection, bone-marrow embolisation, or possibly by atelectasis, opiate administration, or surgery.
Other sickle-cell crises
- Aplastic crises are acute worsenings of the patient's baseline anaemia producing pallor, tachycardia, and fatigue. This crisis is triggered by parvovirus B19, which directly affects erythropoiesis (production of red blood cells). Parvovirus infection nearly completely prevents red blood cell production for 2-3 days. In normal individuals this is of little consequence but the shortened red cell life of sickle-cell patients results in an abrupt, life-threatening situation. Reticulocyte counts drop dramatically during the disease and the rapid turnover of red cells leads to the drop in haemoglobin. Most patients can be managed supportively; some need blood transfusion.
- Splenic sequestration crises are acute, painful enlargements of the spleen. The abdomen becomes bloated and very hard. Management is supportive, sometimes with blood transfusion.
- Hemolytic crises are acute accelerated drop in haemoglobin level. The red blood cells break down at a faster rate. This is particularly common in patients with co-existent G6PD deficiency. Management is supportive, sometimes with blood transfusion.
Complications
Sickle-cell anaemia can lead to various complications, including:
- Overwhelming post-(auto)splenectomy infection (OPSI) is due to functional asplenia, caused by encapsulated organisms such as Streptococcus pneumoniae and Haemophilus influenzae. Daily penicillin prophylaxis is the most commonly used treatment during childhood with some haematologists continuing treatment indefinitely. Patients benefit today from routine vaccination for H. influenzae, S. pneumoniae and Neisseria meningitidis.
- Stroke can result from a progressive vascular narrowing of blood vessels, preventing oxygen from reaching the brain. Cerebral infarction occurs in children, and cerebral hemorrhage in adults.
- Cholelithiasis and cholecystitis (gallstones) may result from excessive bilirubin production and precipitation due to prolonged haemolysis.
- Avascular necrosis (aseptic bone necrosis) of the hip may occur as a result of ischemia.
- Decreased immune reactions due to hyposplenism (malfunctioning of the spleen)
- Priapism and infarction of the penis.
- Osteomyelitis (bacterial bone infection) in individuals with sickle-cell disease is most frequently caused by Salmonella, whereas Staphylococcus is the most common causative organism in the general population.
- Opioid tolerance can occur as a normal, physiologic response to the therapeutic use of opiates. Addiction to opiates occurs no more commonly among individuals with sickle-cell disease than among other individuals treated with opiates for other reasons.
- Acute papillary necrosis in the kidneys.
- Leg ulcers.
- In eyes, background retinopathy, proliferative retinopathy, vitreous haemorrhages and retinal detachments can occur resulting in blindness. Regular annual eye checks are recommended.
- During pregnancy, intrauterine growth retardation, spontaneous abortion and pre-eclampsia are the possibilities.
- Chronic pain: even in the absence of acute vaso-occlusive pain, many patients have chronic pain that is not reported[4]
- Pulmonary hypertension (increased pressure on the pulmonary artery), leading to strain on the right ventricle and a risk of heart failure; typical symptoms are shortness of breath, decreased exercise tolerance and episodes of syncope[5]
- Chronic renal failure - this develops in 4.2% and manifests itself with hypertension (high blood pressure), proteinuria (protein loss in the urine) and worsened anaemia. If it progresses to end-stage renal failure it carries a poor prognosis.[6]
Heterozygotes
The heterozygous form (sickle cell trait) is almost always asymptomatic and the only significant manifestation is the renal concentrating defect presenting with isosthenuria.
Diagnosis
In HbSS, the full blood count reveals haemoglobin levels in the range of 6-8 g/dL with a high reticulocyte count (as the bone marrow compensates for the destruction of sickle cells by producing more red blood cells). In other forms of sickle cell disease, Hb levels tend to be higher. A blood film may show features of hyposplenism (target cells and Howell-Jolly bodies).
Sickling of the red blood cells, on a blood film, can be induced by the addition of sodium metabisulfite. The presence of sickle haemoglobin can also be demonstrated with the "sickle solubility test". A mixture of haemoglobin S (Hb S) in a reducing solution (such as sodium dithionite) gives a turbid appearance while normal Hb gives a clear solution.
Abnormal haemoglobin forms can be detected on haemoglobin electrophoresis, a form of gel electrophoresis on which the various types of haemoglobin move at varying speed. Sickle-cell haemoglobin (HgbS) and haemoglobin C with sickling (HgbSC)—the two most common forms—can be identified from there. The diagnosis can be confirmed with high performance liquid chromatography (HPLC). Genetic testing is rarely performed, as other investigations are highly specific for HbS and HbC.[7]
Pathophysiology
Sickle-cell anaemia is caused by a point mutation in the β-globin chain of haemoglobin, causing the amino acid glutamic acid to be replaced with the hydrophobic amino acid valine at the sixth position. The β-globin gene is found on the short arm of chromosome 11. The association of two wild-type α-globin subunits with two mutant β-globin subunits forms haemoglobin S (HbS). Under low oxygen conditions, the absence of a polar amino acid at position six of the β-globin chain promotes the non-covalent polymerisation (aggregation) of haemoglobin, which distorts red blood cells into a sickle shape and decreases their elasticity.
The loss of red blood cell elasticity is central to the pathophysiology of sickle-cell disease. Normal red blood cells are quite elastic, which allows the cells to deform to pass through capillaries. In sickle-cell disease, low oxygen tension promotes red blood cell sickling and repeated episodes of sickling damage the cell membrane and decrease the cell's elasticity. These cells fail to return to normal shape when normal oxygen tension is restored. Consequently, these rigid blood cells are unable to deform as they pass through narrow capillaries, leading to vessel occlusion and ischaemia.
Genetics

Sickle cell gene mutation probably arose spontaneously in different geographic areas as suggested by restriction endonuclease analysis. These clinically important variants are known as Cameroon, Senegal, Benin, Bantu and Saudi-Asian. Their clinical importance springs from the fact that some of them are associated with higher HbF levels e.g Senegal and Saudi-Asian variants, and tend to have milder disease.[8]
In people heterozygous for HgbS (carriers of sickling haemoglobin), the polymerisation problems are minor. In people homozygous for HgbS, the presence of long chain polymers of HbS distort the shape of the red blood cell, from a smooth donut-like shape to ragged and full of spikes, making it fragile and susceptible to breaking within capillaries. Carriers only have symptoms if they are deprived of oxygen (for example, while climbing a mountain) or while severely dehydrated. Normally these painful crises occur 0.8 times per year per patient. The sickle-cell disease occurs when the seventh amino acid (if we count the initial methionine), glutamic acid is replaced by valine to change its structure and function.

The gene defect is a known mutation of a single nucleotide (see single nucleotide polymorphism - SNP) (A to T) of the β-globin gene, which results in glutamate to be substituted by valine at position 6. Haemoglobin S with this mutation are referred to as HbS, as opposed to the normal adult HbA. The genetic disorder is due to the mutation of a single nucleotide, from a GAG to GTG codon mutation. This is normally a benign mutation, causing no apparent effects on the secondary, tertiary, or quaternary structure of haemoglobin. What it does allow for, under conditions of low oxygen concentration, is the polymerization of the HbS itself. The deoxy form of haemoglobin exposes a hydrophobic patch on the protein between the E and F helices. The hydrophobic residues of the valine at position 6 of the beta chain in haemoglobin are able to associate with the hydrophobic patch, causing haemoglobin S molecules to aggregate and form fibrous precipitates.
The allele responsible for sickle-cell anaemia is autosomal recessive and can be found on the short arm of chromosome 11. A person who receives the defective gene from both father and mother develops the disease; a person who receives one defective and one healthy allele remains healthy, but can pass on the disease and is known as a carrier. If two parents who are carriers have a child, there is a 1-in-4 chance of their child developing the disease and a 1-in-2 chance of their child just being a carrier. Since the gene is incompletely recessive, carriers have a few sickle red blood cells at all times, not enough to cause symptoms, but enough to give resistance to malaria. Because of this, heterozygotes have a higher fitness than either of the homozygotes. This is known as heterozygote advantage.
Due to the evolutionary advantage of the heterozygote, the disease is still prevalent, especially among people with recent ancestry in malaria-stricken areas, such as Africa, the Mediterranean, India and the Middle East.[9]
The Price equation is a simplified mathematical model of the genetic evolution of sickle-cell anaemia.
The malaria parasite has a complex life cycle and spends part of it in red blood cells. In a carrier, the presence of the malaria parasite causes the red blood cell to rupture, making the plasmodium unable to reproduce. Further, the polymerization of Hb affects the ability of the parasite to digest Hb in the first place. Therefore, in areas where malaria is a problem, people's chances of survival actually increase if they carry sickle-cell trait (selection for the heterozygote).
In the USA, where there is no endemic malaria, the prevalence of sickle-cell anaemia amongst African Americans is lower (about 0.25%) than in West Africa (about 4.0%) and is falling. Without endemic malaria from Africa, the condition is purely disadvantageous, and will tend to be bred out of the affected population.

Inheritance
- Sickle-cell conditions are inherited from parents in much the same way as blood type, hair color and texture, eye color and other physical traits.
- The types of haemoglobin a person makes in the red blood cells depend upon what haemoglobin genes are inherited from his parents.
- If one parent has sickle-cell anaemia (SS) and the other has sickle-cell trait (AS), there is a 50% chance (or 1 out of 2) of a child having sickle-cell disease (SS) and a 50% chance of a child having sickle-cell trait (AS).
- When both parents have sickle-cell trait (AS), they have a 25% chance (1 of 4) of a child having sickle-cell disease (SS), as shown in the diagram.
Sickle-cell anaemia appears to be caused by a recessive allele. Two carrier parents have a one in four chance of having a child with the disease. The child will be homozygous recessive.
It has been argued that the allele, although appearing outwardly recessive, is in fact co-dominant, due to the resistance to a malaria which is obtained by those of the AS genotype. Since a separate phenotype from that of Normal (AA) has therefore been expressed, it is impossible to argue that the S allele is homozygous recessive.
Treatment
Painful (vaso-occlusive) crises
Most people with sickle-cell disease have intensely painful episodes called vaso-occlusive crises. The frequency, severity, and duration of these crises, however, vary tremendously. Painful crises are treated symptomatically with analgesics; pain management requires opioid administration at regular intervals until the crisis has settled. For milder crises a subgroup of patients manage on NSAIDs (such as diclofenac or naproxen). For more severe crises most patients require inpatient management for intravenous opioids; patient-controlled analgesia (PCA) devices are commonly used in this setting. Diphenhydramine is also an effective agent that is frequently prescribed by doctors in order to help control any itching associated with the use of opioids.
Folic acid and penicillin
Children born with sickle cell disease will undergo close observation by the pediatrician and will require management by a hemotologist to assure they remain healthy. These patients will take folic acid 1mg daily for life. From the age of birth to 5 years of age they will also have to take penicillin daily, due to the immature immune system which makes them more prone to early childhood illnesses.
Acute chest crises
Management is similar to vaso-occlusive crises with the addition of antibiotics (usually a quinolone or macrolide, since wall-deficient ["atypical"] bacteria are thought to contribute to the syndrome),[10] oxygen supplementation for hypoxia, and close observation. Should the pulmonary infiltrate worsen or the oxygen requirements increase, simple blood transfusion or exchange transfusion is indicated. The latter involves the exchange of a significant portion of the patients red cell mass for normal red cells, which decreases the percent haemoglobin S in the patient's blood. Blood transfusion not a routine and automatic occurrence in these cases due to iron overload (hemosiderosis).
Hydroxyurea
The first approved drug for the causative treatment of sickle-cell anaemia, hydroxyurea, was shown to decrease the number and severity of attacks in a study in 1995 (Charache et al)[11] and shown to possibly increase survival time in a study in 2003 (Steinberg et al).[12] This is achieved, in part, by reactivating fetal haemoglobin production in place of the haemoglobin S that causes sickle-cell anaemia. Hydroxyurea had previously been used as a chemotherapy agent, and there is some concern that long-term use may be harmful, but this risk has been shown to be either absent or very small and it is likely that the benefits outweigh the risks.[13]
Bone marrow transplants
Bone marrow transplants have proven to be effective in children.
Future treatments
Various approaches are being sought for preventing sickling episodes as well as for the complications of sickle-cell disease. Other ways to modify hemoglobin switching are being investigated, including the use of phytochemicals such as nicosan. Gene therapy is under investigation.
Situation of carriers
People who are known carriers of the disease often undergo genetic counseling before they have a child. A test to see if an unborn child has the disease takes either a blood sample from the fetus or a sample of amniotic fluid. Since taking a blood sample from a fetus has risks, the latter test is usually used.
After the mutation responsible for this disease was discovered in 1979, the U.S. Air Force required African American applicants to test for the mutation. It dismissed 143 applicants because they were carriers, even though none of them had the condition. It eventually withdrew the requirement, but only after a trainee filed a lawsuit.
History
This collection of clinical findings was unknown until the explanation of the sickle cells in 1904 by the Chicago cardiologist and professor of medicine James B. Herrick (1861-1954) whose intern Ernest Edward Irons (1877-1959) found "peculiar elongated and sickle shaped" cells in the blood of Walter Clement Noel, a 20 year old first year dental student from Grenada after Noel was admitted to the Chicago Presbyterian Hospital in December 1904 suffering from anaemia. Noel was readmitted several times over the next three years for "muscular rheumatism" and "bilious attacks". Noel completed his studies and returned to the capital of Grenada (St. George's) to practice dentistry. He died of pneumonia in 1916 and is buried in the Catholic cemetery at Sauteurs in the north of Grenada.[14]
The disease was named "sickle-cell anaemia" by Vernon Mason in 1922. In retrospect some elements of the disease had been recognized earlier: a paper in the Southern Journal of Medical Pharmacology in 1846 described the absence of a spleen in the autopsy of a runaway slave. The African medical literature reported this condition in the 1870s where it was known locally as ogbanjes ("children who come and go") because of the very high infant mortality rate caused by this condition. A history of the condition tracked reports back to 1670 in one Ghanaian family.[15] Also, the practice of using tar soap to cover blemishes caused by sickle-cell sores was prevalent in the African American community.
Linus Pauling and colleagues were the first, in 1949, to demonstrate that sickle cell disease occurs as a result of an abnormality in the haemoglobin molecule. This was the first time a genetic disease was linked to a mutation of a specific protein, a milestone in the history of molecular biology.
The origin of the mutation that led to the sickle-cell gene was initially thought to be in the Arabian peninsula, spreading to Asia and Africa. It is now known, from evaluation of chromosome structures, that there have been at least four independent mutational events, three in Africa and a fourth in either Saudi Arabia or central India.[16] These independent events occurred between 3,000 and 6,000 generations ago, approximately 70-150,000 years.
Notes
- ↑ Platt OS, Brambilla DJ, Rosse WF, et al (June 1994). "Mortality in sickle cell disease. Life expectancy and risk factors for early death". N. Engl. J. Med. 330 (23): 1639–44. doi:. PMID 7993409. http://content.nejm.org/cgi/content/full/330/23/1639.
- ↑ Pearson H. "Sickle cell anaemia and severe infections due to encapsulated bacteria". J Infect Dis 136 Suppl: S25–30. PMID 330779.
- ↑ Wong W, Powars D, Chan L, Hiti A, Johnson C, Overturf G (1992). "Polysaccharide encapsulated bacterial infection in sickle cell anaemia: a thirty year epidemiologic experience". Am J Hematol 39 (3): 176–82. doi:. PMID 1546714.
- ↑ Smith WR, Penberthy LT, Bovbjerg VE, et al (2008). "Daily assessment of pain in adults with sickle cell disease". Ann. Intern. Med. 148 (2): 94–101. PMID 18195334.
- ↑ Gladwin MT, Sachdev V, Jison ML, et al (2004). "Pulmonary hypertension as a risk factor for death in patients with sickle cell disease". N. Engl. J. Med. 350 (9): 886–95. doi:. PMID 14985486. http://content.nejm.org/cgi/content/full/350/9/886.
- ↑ Powars DR, Elliott-Mills DD, Chan L, et al (1991). "Chronic renal failure in sickle cell disease: risk factors, clinical course, and mortality". Ann. Intern. Med. 115 (8): 614–20. PMID 1892333.
- ↑ Clarke GM, Higgins TN (2000). "Laboratory investigation of hemoglobinopathies and thalassemias: review and update". Clin. Chem. 46 (8 Pt 2): 1284–90. PMID 10926923. http://www.clinchem.org/cgi/content/full/46/8/1284.
- ↑ Green NS, Fabry ME, Kaptue-Noche L, Nagel RL (1993). "Senegal haplotype is associated with higher HbF than Benin and Cameroon haplotypes in African children with sickle cell anemia". Am. J. Hematol. 44 (2): 145–6. doi:. PMID 7505527.
- ↑ Kwiatkowski DP (2005). "How malaria has affected the human genome and what human genetics can teach us about malaria". Am. J. Hum. Genet. 77 (2): 171–92. doi:. PMID 16001361. Full text at PMC: 1224522
- ↑ Aldrich TK, Nagel RL. (1998). "Pulmonary Complications of Sickle Cell Disease.". in Bone RC et al., editors. Pulmonary and Critical Care Medicine (6th edition ed.). St. Louis: Mosby. pp. pp.1–10. ISBN 0-81511-371-4.
- ↑ Charache S, Terrin ML, Moore RD, et al (1995). "Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia". N. Engl. J. Med. 332 (20): 1317–22. doi:. PMID 7715639.
- ↑ Steinberg MH, Barton F, Castro O, et al (2003). "Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: risks and benefits up to 9 years of treatment". JAMA 289 (13): 1645–51. doi:. PMID 12672732. http://jama.ama-assn.org/cgi/content/full/289/13/1645.
- ↑ Platt OS (2008). "Hydroxyurea for the treatment of sickle cell anemia". N. Engl. J. Med. 358 (13): 1362–9. doi:. PMID 18367739.
- ↑ Savitt TL, Goldberg MF (1989). "Herrick's 1910 case report of sickle cell anemia. The rest of the story". JAMA 261 (2): 266–71. doi:. PMID 2642320.
- ↑ Konotey-Ahulu FID. Effect of environment on sickle cell disease in West Africa: epidemiologic and clinical considerations. In: Sickle Cell Disease, Diagnosis, Management, Education and Research. Abramson H, Bertles JF, Wethers DL, eds. CV Mosby Co, St. Louis. 1973; 20; cited in Desai, D. V.; Hiren Dhanani (2004). "Sickle Cell Disease: History And Origin". The Internet Journal of Haematology 1 (2). ISSN 1540-2649. http://www.ispub.com/ostia/index.php?xmlFilePath=journals/ijhe/vol1n2/sickle.xml.
- ↑ Desai, D. V.; Hiren Dhanani (2004). "Sickle Cell Disease: History And Origin". The Internet Journal of Haematology 1 (2). ISSN 1540-2649. http://www.ispub.com/ostia/index.php?xmlFilePath=journals/ijhe/vol1n2/sickle.xml.
Further reading
- Chestnut, D. (1994). Perceptions of ethnic and cultural factors in the delivery of services in the treatment of sickle cell disease. Journal of Health and Social Policy, 5(3/4), 236.
- Jurmain, Bruce.; Lynn Kilgore, Wenda Trevathan (2005). ~~Introduction to Physical Anthropology~~. ISBN 0-495-18779-8
External links
- Sickle cell at the Open Directory Project
- Sickle cell (kids) at the Open Directory Project
- Sickle Cell Disease Association of America
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||