Phenylalanine
- Phe redirects here. For the BitTorrent feature, see PHE. For the constellation, see Phoenix (constellation).
| Phenylalanine | |
|---|---|
 |
 |
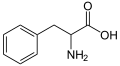
| IUPAC name | 2-Amino-3-phenyl-propanoic acid |
| Identifiers | |
| CAS number | 150-30-1 |
| PubChem | |
| SMILES |
|
| ChemSpider ID | |
| Properties | |
| Molecular formula | C9H11NO2 |
| Molar mass | 165.19 g mol−1 |
| Except where noted otherwise, data are given for materials in their standard state (at 25 °C, 100 kPa) Infobox references |
|
Phenylalanine (abbreviated as Phe or F)[1] is an α-amino acid with the formula HO2CCH(NH2)CH2C6H5, which is found naturally in the breast milk of mammals and manufactured for food and drink products and are also sold as nutritional supplements for their reputed analgesic and antidepressant effects. Phenylalanine is structurally closely related to dopamine, epinepherine (adrenaline) and tyrosine.
This essential amino acid is classified as nonpolar because of the hydrophobic nature of the benzyl side chain. The codons for L-phenylalanine are UUU and UUC. It is a white, powdery solid. L-Phenylalanine (LPA) is an electrically-neutral amino acid, one of the twenty common amino acids used to biochemically form proteins, coded for by DNA.
Contents |
Biosynthesis
Breast milk from mammals is rich in phenylalanine. It is also produced by plants and most microorganisms from prephenate, an intermediate on the shikimate pathway.[2]
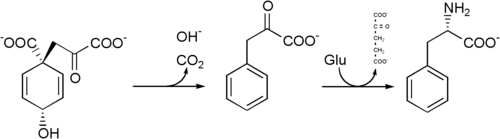
Prephenate is decarboxylated with loss of the hydroxyl group to give phenylpyruvate. This species is transaminated using glutamate as the nitrogen source to give phenylalanine and α-ketoglutarate.
Other biological roles
L-phenylalanine can also be converted into L-tyrosine, another one of the DNA-encoded amino acids. L-tyrosine in turn is converted into L-DOPA, which is further converted into dopamine, norepinephrine (noradrenaline), and epinephrine (adrenaline). The latter three are known as the catecholamines.
Phenylalanine uses the same active transport channel as tryptophan to cross the blood-brain barrier, and, in large quantities, interferes with the production of serotonin.
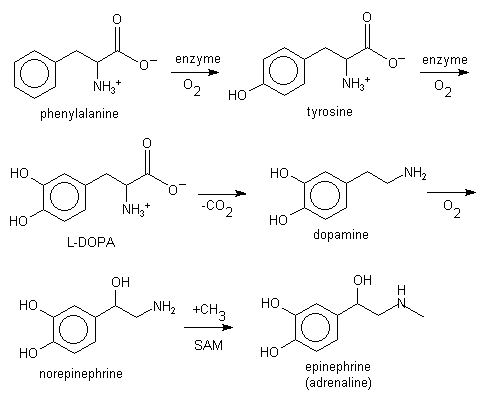
Lignin is derived from phenylalanine and from tyrosine. Phenylalanine is converted to cinnamic acid by the enzyme phenylalanine ammonia lyase.[2]
Phenylketonuria
The genetic disorder phenylketonuria (PKU) is the inability to metabolize phenylalanine. Individuals with this disorder are known as "phenylketonurics" and must abstain from consumption of too much phenylalanine. This dietary restriction also applies to pregnant women with hyperphenylalanine (high levels of phenylalanine in blood) because they do not properly metabolize the amino acid phenylalanine. Persons suffering from PKU must monitor their intake of protein to control the buildup of phenylalanine as their bodies convert protein into its component amino acids.
A non food source of phenylalanine is the artificial sweetener aspartame. This compound, sold under the trade names "Equal" and "NutraSweet", is metabolized by the body into several chemical byproducts including phenylalanine. The breakdown problems phenylketonurics have with protein and the attendant build up of phenylalanine in the body also occurs with the ingestion of aspartame, although to a lesser degree. Accordingly, all products in Australia, the U.S. and Canada that contain aspartame must be labeled: "Phenylketonurics: Contains phenylalanine." In the UK, foods containing aspartame must carry ingredients panels that refer to the presence of "aspartame or E951" [3] and they must be labeled with a warning "Contains a source of phenylalanine." These warnings are specifically placed to aid individuals who suffer from PKU so that they can avoid such foods.
Geneticists have recently sequenced the genome of macaques. Their investigations have found "some instances where the normal form of the macaque protein looks like the diseased human protein" including markers for PKU.[4]
D- and DL-phenylalanine
The unnatural stereoisomer D-phenylalanine (DPA) can be produced by conventional organic synthesis, either as a single enantiomer or as a component of the racemic mixture. It does not participate in protein biosynthesis although it is found in proteins in small amounts - particularly aged proteins and food proteins that have been processed. The biological functions of D-amino acids remain unclear although some, such as D-phenylalanine, may have pharmacological activity.
DL-Phenylalanine is marketed as a nutritional supplement for its supposed analgesic and antidepressant activities. The reputed analgesic activity of DL-phenylalanine may be explained by the possible blockage by D-phenylalanine of enkephalin degradation by the enzyme carboxypeptidase A.[5] The mechanism of DL-phenylalanine's supposed antidepressant activity may be accounted for by the precursor role of L-phenylalanine in the synthesis of the neurotransmitters, norepinephrine and dopamine. Elevated brain levels of norepinephrine and dopamine are thought to have an antidepressant effect. Following ingestion, D-Phenylalanine is absorbed from the small intestine and transported to the liver via the portal circulation. A small amount of D-phenylalanine appears to be converted to L-phenylalanine. D-Phenylalanine is distributed to the various tissues of the body via the systemic circulation. It appears to cross the blood-brain barrier less efficienciently than L-phenylalanine, and so a small amount of an ingested dose of D-phenylalanine is not absorbed but excreted in the urine.
History
The genetic codon for phenylalanine was first discovered by J. Heinrich Matthaei and Marshall W. Nirenberg in 1961. They showed that by using m-RNA to insert multiple uracil repeats into the bacterium E. coli, the bacterium produced a new protein consisting solely of repeated phenylalanine amino acids. This discovery lead to the determination of the relationship between RNA and amino acids, which was foundational to the understanding of the Genetic Code.
References
- ↑ IUPAC-IUBMB Joint Commission on Biochemical Nomenclature. "Nomenclature and Symbolism for Amino Acids and Peptides". Recommendations on Organic & Biochemical Nomenclature, Symbols & Terminology etc. Retrieved on 2007-05-17.
- ↑ 2.0 2.1 Nelson, D. L.; Cox, M. M. "Lehninger, Principles of Biochemistry" 3rd Ed. Worth Publishing: New York, 2000. ISBN 1-57259-153-6.
- ↑ Aspartame, Food Standards Agency
- ↑ Scientists decode macaque genome, BBC News, 13 April 2007
- ↑ Christianson DW, Mangani S, Shoham G, Lipscomb WN. "Binding of D-phenylalanine and D-tyrosine to carboxypeptidase A." Journal of Biological Chemistry 1989 Aug 5;264(22):12849-53. PMID: 2568989.
External links
|
||||||||||||||