Hemoglobin
|
Hemoglobin, human, adult
(heterotetramer, (αβ)2) |
||
 |
||
| Structure of human hemoglobin. The protein's α and β subunits are in red and blue, and the iron-containing heme groups in green. From PDB 1GZX Proteopedia Hemoglobin | ||
| - | ||
| Protein type | metalloprotein, globulin | |
| Function | oxygen-transport | |
| Cofactor(s) | heme (4) | |
| - | ||
| Subunit Name |
Gene | Chromosomal Locus |
| Hb α1 | HBA1 | Chromosome 16p13.3 |
| Hb α2 | HBA2 | Chromosome 16p13.3 |
| Hb β | HBB | Chromosome 11p15.5 |
Hemoglobin (also spelled haemoglobin and abbreviated Hb or Hgb) is the iron-containing oxygen-transport metalloprotein in the red blood cells of vertebrates.[1]
In mammals, the protein makes up about 97% of the red blood cell’s dry content, and around 35% of the total content (including water). Hemoglobin transports oxygen from the lungs or gills to the rest of the body, such as to the muscles, where it releases the oxygen for cell use. It also has a variety of other roles of gas transport and effect-modulation which vary from species to species, and are quite diverse in some invertebrates.
Discovery
The oxygen-carrying protein hemoglobin was discovered by Otto Funke in 1851.[2] In that year he published a series of articles in which he described growing hemoglobin crystals by successively diluting red blood cells with a solvent such as pure water, alcohol or ether, followed by slow evaporation of the solvent from the resulting protein solution.[3]
Hemoglobin’s reversible oxygenation was described a few years later by Felix Hoppe-Seyler.[4] In 1959 Max Perutz determined the molecular structure of hemoglobin by X-ray crystallography.[5][6] This work resulted in his sharing with John Kendrew the 1962 Nobel Prize in Chemistry.
The role of hemoglobin in the blood was elucidated by physiologist Claude Bernard. The name hemoglobin is the concatenation of heme and globin, reflecting the fact that each subunit of hemoglobin is a globular protein with an embedded heme (or haem) group. Each heme group contains one iron atom, that can bind one oxygen molecule through ion-induced dipole forces. The most common type of hemoglobin in mammals contains four such subunits.
Genetics
Mutations in the genes for the hemoglobin protein in a species result in hemoglobin variants,[7][8] some of which cause a group of hereditary diseases termed the hemoglobinopathies in humans. The best known is sickle-cell disease, which was the first human disease whose mechanism was understood at the molecular level. A (mostly) separate set of diseases called thalassemias involves underproduction of normal and sometimes abnormal hemoglobins, through problems and mutations in globin gene regulation. These diseases also often produce anemia.[9]
The chemical formulas of hemoglobins vary widely across species, and even (through common mutations) slightly among subgroups of humans.
Synthesis
Hemoglobin (Hb) is synthesized in a complex series of steps. The heme part is synthesized in a series of steps in the mitochondria and the cytosol of immature red blood cells, while the globin protein parts are synthesized by ribosomes in the cytosol.[10] Production of Hb continues in the cell throughout its early development from the proerythroblast to the reticulocyte in the bone marrow. At this point, the nucleus is lost in mammalian red blood cells, but not in birds and many other species. Even after the loss of the nucleus in mammals, residual ribosomal RNA allows further synthesis of Hb until the reticulocyte loses its RNA soon after entering the vasculature (this hemoglobin-synthetic RNA in fact gives the reticulocyte its reticulated appearance and name).
Structure

In most humans, the hemoglobin molecule is an assembly of four globular protein subunits. Each subunit is composed of a protein chain tightly associated with a non-protein heme group. Each protein chain arranges into a set of alpha-helix structural segments connected together in a globin fold arrangement, so called because this arrangement is the same folding motif used in other heme/globin proteins such as myoglobin.[11][12] This folding pattern contains a pocket which strongly binds the heme group.
A heme group consists of an iron (Fe) ion (charged atom) held in a heterocyclic ring, known as a porphyrin. The iron ion, which is the site of oxygen binding, coordinates with the four nitrogens in the center of the ring, which all lie in one plane. The iron is also bound strongly to the globular protein via the imidazole ring of the F8 histidine residue below the porphyrin ring. A sixth position can reversibly bind oxygen, completing the octahedral group of six ligands. Oxygen binds in an "end-on bent" geometry where one oxygen atom binds Fe and the other protrudes at an angle. When oxygen is not bound, a very weakly bonded water molecule fills the site, forming a distorted octahedron.
The iron ion may either be in the Fe2+ or Fe3+ state, but ferrihemoglobin (methemoglobin) (Fe3+) cannot bind oxygen.[13] In binding, oxygen temporarily oxidizes Fe to (Fe3+), so iron must exist in the +2 oxidation state in order to bind oxygen. The enzyme methemoglobin reductase reactivates hemoglobin found in the inactive (Fe3+) state by reducing the iron center.
In adult humans, the most common hemoglobin type is a tetramer (which contains 4 subunit proteins) called hemoglobin A, consisting of two α and two β subunits non-covalently bound, each made of 141 and 146 amino acid residues, respectively. This is denoted as α2β2. The subunits are structurally similar and about the same size. Each subunit has a molecular weight of about 17,000 daltons, for a total molecular weight of the tetramer of about 68,000 daltons. Hemoglobin A is the most intensively studied of the hemoglobin molecules.
The four polypeptide chains are bound to each other by salt bridges, hydrogen bonds, and hydrophobic interactions. There are two kinds of contacts between the α and β chains: α1β1 and α2β2.
Oxyhemoglobin is formed during respiration when oxygen binds to the heme component of the protein hemoglobin in red blood cells. This process occurs in the pulmonary capillaries adjacent to the alveoli of the lungs. The oxygen then travels through the blood stream to be dropped off at cells where it is utilized in aerobic glycolysis and in the production of ATP by the process of oxidative phosphorylation. It does not, however, help to counteract a decrease in blood pH. Ventilation, or breathing, may reverse this condition by removal of carbon dioxide, thus causing a shift up in pH.[14]
Deoxyhemoglobin is the form of hemoglobin without the bound oxygen. The absorption spectra of oxyhemoglobin and deoxyhemoglobin differ. The oxyhemoglobin has significantly lower absorption of the 660 nm wavelength than deoxyhemoglobin, while at 940 nm its absorption is slightly higher. This difference is used for measurement of the amount of oxygen in patient's blood by an instrument called pulse oximeter.
Iron's oxidation state in oxyhemoglobin
Assigning oxygenated hemoglobin's oxidation state is difficult because oxyhemoglobin (Hb-O2), by experimental measurement, is diamagnetic (no net unpaired electrons), yet the low-energy electron configurations in both oxygen and iron are paramagnetic (suggesting at least one unpaired electron in the complex). The low-energy forms are these:
- Triplet oxygen, the lowest energy molecular oxygen species, has two unpaired electrons in antibonding π* molecular orbitals.
- Iron(II) tends to exist in a high-spin configuration where unpaired electrons exist in Eg antibonding orbitals.
- Iron(III) has an odd number of electrons, and thus must have one or more unpaired electrons, in any energy state.
All of these structures are paramagnetic (have unpaired electrons), not diamagnetic. Thus, a non-intuitive distribution of electrons in the combination of iron and oxygen must exist, to explain the observed diamagnetism and no unpaired electrons.
The three logical possibilities to produce diamagnetic (no net spin) Hb-O2 are:
- Low-spin Fe2+ binds to singlet oxygen. Both low-spin iron and singlet oxygen are diamagnetic. However, the singlet form of oxygen is the higher-energy form of the molecule.
- Low-spin Fe3+ binds to .O2- (the superoxide ion) and the two unpaired electrons couple antiferromagnetically, giving diamagnetic properties.
- Low-spin Fe4+ binds to peroxide, O22-. Both are diamagnetic.
X-ray photoelectron spectroscopy suggests iron has an oxidation state of approximately 3.2 and infrared stretching frequencies of the O-O bond suggests a bond length fitting with superoxide (a bond order of about 1.6, with superoxide being 1.5). The nearest formal oxidation state of iron in Hb-O2 is thus the +3 state, with oxygen in the -1 state (as superoxide .O2-). The diamagnetism in this configuration arises from the single unpaired electron on superoxide aligning antiferromagnetically from the single unpaired electron on iron, to give no net spin to the entire configuration, in accordance with experiment.[15]
The second choice being correct is not surprising because singlet oxygen (possibility #1) and large separations of charge (possibility #3) are both unfavorably high-energy states. Iron's shift to a higher oxidation state in Hb-O2 decreases the atom's size, and allows it into the plane of the porphyrin ring, pulling on the coordinated histidine residue and initiating the allosteric changes seen in the globulins.
Early postulates by bio-inorganic chemists claimed that possibility #1 (above) was correct and that iron should exist in oxidation state II. This seemed particularly likely since the iron oxidation state III as methemoglobin, when not accompanied by superoxide .O2- to "hold" the oxidation electron, was known to render hemoglobin incapable of binding normal triplet O2 as it occurs in the air. It was thus assumed that iron remained as Fe(II) when oxygen gas was bound in the lungs. The iron chemistry in this previous classical model was elegant, but the required presence of the required diamagnetic high-energy singlet oxygen was never explained. It was classically argued that the binding of an oxygen molecule placed high-spin iron(II) in an octahedral field of strong-field ligands; this change in field would increase the crystal field splitting energy, causing iron's electrons to pair into the low-spin configuration, which would be diamagnetic in Fe(II). This forced low-spin pairing is indeed thought to happen in iron when oxygen binds, but is not enough to explain iron's change in size. Extraction of an additional electron from iron by oxygen is required to explain both iron's smaller size and observed increased oxidation state, and oxygen's weaker bond.
It should be noted that the assignment of a whole-number oxidation state is a formalism, as the covalent bonds are not required to have perfect bond orders involving whole electron-transfer. Thus, all three models for paramagnetic Hb-O2 may contribute to some small degree (by resonance) to the actual electronic configuration of Hb-O2. However, the model of iron in Hb-O2 being Fe(III) is more correct than the classical idea that it remains Fe(II).
Ligand binding
Besides the oxygen ligand which binds to hemoglobin in a cooperative manner, hemoglobin ligands also include competitive inhibitors such as carbon monoxide (CO) and allosteric ligands such as carbon dioxide (CO2).
Cooperative


When oxygen binds to the iron complex, it causes the iron atom to move back toward the center of the plane of the porphyrin ring (see moving diagram). At the same time, the imidazole side chain of the histidine residue interacting at the other pole of the iron is pulled toward the porphyrin ring. This interaction forces the plane of the ring sideways toward the outside of the tetramer, and also induces a strain in the protein helix containing the histidine as it moves nearer to the iron atom. This strain is transmitted to the remaining three monomers in the tetramer where it induces a similar conformational change in the other heme sites such that binding of oxygen to these site becomes easier.
In the tetrameric form of normal adult hemoglobin, the binding of oxygen is thus a cooperative process. The binding affinity of hemoglobin for oxygen is increased by the oxygen saturation of the molecule, with the first oxygens bound influencing the shape of the binding sites for the next oxygens, in a way favorable for binding. This positive cooperative binding is achieved through steric conformational changes of the hemoglobin protein complex as discussed above, i.e. when one subunit protein in hemoglobin becomes oxygenated, this induces a conformational or structural change in the whole complex, causing the other subunits to gain an increased affinity for oxygen. As a consequence, the oxygen binding curve of hemoglobin is sigmoidal, or S-shaped, as opposed to the normal hyperbolic curve associated with noncooperative binding.
Competitive
Hemoglobin's oxygen-binding capacity is decreased in the presence of carbon monoxide because both gases compete for the same binding sites on hemoglobin, carbon monoxide binding preferentially in place of oxygen.
The binding of oxygen is affected by molecules such as carbon monoxide (CO) (for example from tobacco smoking, car exhuast and incomplete combustion in furnaces). CO competes with oxygen at the heme binding site. Hemoglobin binding affinity for CO is 200 times greater than its affinity for oxygen, meaning that small amounts of CO dramatically reduce hemoglobin's ability to transport oxygen. When hemoglobin combines with CO, it forms a very bright red compound called carboxyhemoglobin. When inspired air contains CO levels as low as 0.02%, headache and nausea occur; if the CO concentration is increased to 0.1%, unconsciousness will follow. In heavy smokers, up to 20% of the oxygen-active sites can be blocked by CO.
In similar fashion, hemoglobin also has competitive binding affinity for cyanide (CN-), sulfur monoxide (SO), nitrogen dioxide (NO2), and sulfide(S2-), including hydrogen sulfide (H2S). All of these bind to iron in heme without changing its oxidation state, but they nevertheless inhibit oxygen-binding, causing grave toxicity.
The iron atom in the heme group must be in the ferrous (Fe2+) oxidation state to support oxygen and other gases' binding and transport. Oxidation to the ferric (Fe3+) state converts hemoglobin into hemiglobin or methemoglobin (pronounced "MET-hemoglobin"), which cannot bind oxygen. Hemoglobin in normal red blood cells is protected by a reduction system to keep this from happening. Nitrogen dioxide and nitrous oxide are capable of converting a small fraction of hemoglobin to methemoglobin; however, this is not usually of medical importance (nitrogen dioxide is poisonous by other mechanisms, and nitrous oxide is routinely used in surgical anesthesia in most people without undue methemoglobin buildup).
Allosteric
Carbon dioxide occupies a different binding site on the hemoglobin. Carbon dioxide is more readily dissolved in deoxygenated blood, facilitating its removal from the body after the oxygen has been released to tissues undergoing metabolism. This increased affinity for carbon dioxide by the venous blood is known as the Haldane effect. Through the enzyme carbonic anhydrase, carbon dioxide reacts with water to give carbonic acid, which decomposes into bicarbonate and protons:
- CO2 + H2O → H2CO3 → HCO3- + H+
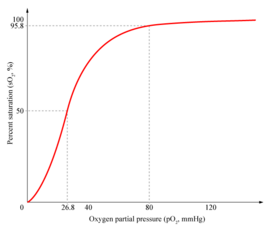
Hence blood with high carbon dioxide levels is also lower in pH (more acidic). Hemoglobin can bind protons and carbon dioxide which causes a conformational change in the protein and facilitates the release of oxygen. Protons bind at various places along the protein, and carbon dioxide also binds at the alpha-amino group forming carbamate. This decrease in hemoglobin's affinity for oxygen by the binding of carbon dioxide and acid is known as the Bohr effect (shifts the O2-saturation curve to the right). Conversely, when the carbon dioxide levels in the blood decrease (i.e., in the lung capillaries), carbon dioxide and protons are released from hemoglobin, increasing the oxygen affinity of the protein.
In people acclimated to high altitudes, the concentration of 2,3-Bisphosphoglycerate (2,3-BPG) in the blood is increased, which allows these individuals to deliver a larger amount of oxygen to tissues under conditions of lower oxygen tension. This phenomenon, where molecule Y affects the binding of molecule X to a transport molecule Z, is called a heterotropic allosteric effect.
A variant hemoglobin, called fetal hemoglobin (HbF, α2γ2), is found in the developing fetus, and binds oxygen with greater affinity than adult hemoglobin. This means that the oxygen binding curve for fetal hemoglobin is left-shifted (i.e., a higher percentage of hemoglobin has oxygen bound to it at lower oxygen tension), in comparison to that of adult hemoglobin. As a result, fetal blood in the placenta is able to take oxygen from maternal blood.
Hemoglobin also carries nitric oxide in the globin part of the molecule. This improves oxygen delivery in the periphery and contributes to the control of respiration. NO binds reversibly to a specific cysteine residue in globin; the binding depends on the state (R or T) of the hemoglobin. The resulting S-nitrosylated hemoglobin influences various NO-related activities such as the control of vascular resistance, blood pressure and respiration. NO is released not in the cytoplasm of erythrocytes but is transported by an anion exchanger called AE1 out of them.[16]
Types in humans
|
hemoglobin, alpha 1
|
|
| Identifiers | |
| Symbol | HBA1 |
| Entrez | 3039 |
| HUGO | 4823 |
| OMIM | 141800 |
| RefSeq | NM_000558 |
| UniProt | P69905 |
| Other data | |
| Locus | Chr. 16 p13.3 |
|
hemoglobin, alpha 2
|
|
| Identifiers | |
| Symbol | HBA2 |
| Entrez | 3040 |
| HUGO | 4824 |
| OMIM | 141850 |
| RefSeq | NM_000517 |
| UniProt | P69905 |
| Other data | |
| Locus | Chr. 16 p13.3 |
|
hemoglobin, beta
|
|
| Identifiers | |
| Symbol | HBB |
| Entrez | 3043 |
| HUGO | 4827 |
| OMIM | 141900 |
| RefSeq | NM_000518 |
| UniProt | P68871 |
| Other data | |
| Locus | Chr. 11 p15.5 |
Hemoglobin variants are a part of the normal embryonic and fetal development, but may also be pathologic mutant forms of hemoglobin in a population, caused by variations in genetics. Some well-known hemoglobin such variants such as sickle-cell anemia are responsible for diseases, and are considered hemoglobinopathies. Other variants cause no detectable pathology, and are thus considered non-pathological variants.[17][18]
In the embryo:
- Gower 1 (ζ2ε2)
- Gower 2 (α2ε2) (PDB 1A9W)
- Hemoglobin Portland (ζ2γ2)
In the fetus:
- Hemoglobin F (α2γ2) (PDB 1FDH)
In adults:
- Hemoglobin A (α2β2) (PDB 1BZ0) - The most common with a normal amount over 95%
- Hemoglobin A2 (α2δ2) - δ chain synthesis begins late in the third trimester and in adults, it has a normal range of 1.5-3.5%
- Hemoglobin F (α2γ2) - In adults Hemoglobin F is restricted to a limited population of red cells called F-cells. However, the level of Hb F can be elevated in persons with sickle-cell disease.
Variant forms which cause disease:
- Hemoglobin S (α2βS2) - A variant form of hemoglobin found in people with sickle cell disease. There is a variation in the β-chain gene, causing a change in the properties of hemoblobin which results in sickling of red blood cells.
- Hemoglobin C (α2βC2) - Another variant due to a variation in the β-chain gene. This variant causes a mild chronic hemolytic anemia.
Degradation in vertebrate animals
When red cells reach the end of their life due to aging or defects, they are broken down, the hemoglobin molecule is broken up and the iron gets recycled. When the porphyrin ring is broken up, the fragments are normally secreted in the bile by the liver. This process also produces one molecule of carbon monoxide for every molecule of heme degraded.[19] This is one of the few natural sources of carbon monoxide production in the human body, and is responsible for the normal blood levels of carbon monoxide even in people breathing pure air. The other major final product of heme degradation is bilirubin. Increased levels of this chemical are detected in the blood if red cells are being destroyed more rapidly than usual. Improperly degraded hemoglobin protein or hemoglobin that has been released from the blood cells too rapidly can clog small blood vessels, especially the delicate blood filtering vessels of the kidneys, causing kidney damage.
Role in disease

Decrease of hemoglobin, with or without an absolute decrease of red blood cells, leads to symptoms of anemia. Anemia has many different causes, although iron deficiency and its resultant iron deficiency anemia are the most common causes in the Western world. As absence of iron decreases heme synthesis, red blood cells in iron deficiency anemia are hypochromic (lacking the red hemoglobin pigment) and microcytic (smaller than normal). Other anemias are rarer. In hemolysis (accelerated breakdown of red blood cells), associated jaundice is caused by the hemoglobin metabolite bilirubin, and the circulating hemoglobin can cause renal failure.
Some mutations in the globin chain are associated with the hemoglobinopathies, such as sickle-cell disease and thalassemia. Other mutations, as discussed at the beginning of the article, are benign and are referred to merely as hemoglobin variants.
There is a group of genetic disorders, known as the porphyrias that are characterized by errors in metabolic pathways of heme synthesis. King George III of the United Kingdom was probably the most famous porphyria sufferer.
To a small extent, hemoglobin A slowly combines with glucose at the terminal valine (an alpha aminoacid) of each β chain. The resulting molecule is often referred to as Hb A1c. As the concentration of glucose in the blood increases, the percentage of Hb A that turns into Hb A1c increases. In diabetics whose glucose usually runs high, the percent Hb A1c also runs high. Because of the slow rate of Hb A combination with glucose, the Hb A1c percentage is representative of glucose level in the blood averaged over a longer time (the half-life of red blood cells, which is typically 50-55 days).
Elevated levels of hemoglobin are associated with increased numbers or sizes of red blood cells, called polycythemia. This elevation may be caused by congenital heart disease, cor pulmonale, pulmonary fibrosis, too much erythropoietin, or polycythemia vera.[20]
Diagnostic uses
Hemoglobin concentration measurement is among the most commonly performed blood tests, usually as part of a complete blood count. For example it is typically tested before blood donation. Results are reported in g/L, g/dL or mol/L. 1 g/dL equals about 0.6206 mmol/L. Normal levels are:
- Men: 13.5 to 16.5 g/dl
- Women: 12.1 to 15.1 g/dl
- Children: 11 to 16 g/dl
- Pregnant women: 11 to 12 g/dl [21]
If the concentration is below normal, this is called anemia. Anemias are classified by the size of red blood cells, the cells which contain hemoglobin in vertebrates. The anemia is called "microcytic" if red cells are small, "macrocytic" if they are large, and "normocytic" otherwise.
Hematocrit, the proportion of blood volume occupied by red blood cells, is typically about three times the hemoglobin level. For example, if the hemoglobin is measured at 17, that compares with a hematocrit of 51.[22]
Long-term control of blood sugar concentration can be measured by the concentration of Hb A1c. Measuring it directly would require many samples because blood sugar levels vary widely through the day. Hb A1c is the product of the irreversible reaction of hemoglobin A with glucose. A higher glucose concentration results in more Hb A1c. Because the reaction is slow, the Hb A1c proportion represents glucose level in blood averaged over the half-life of red blood cells, is typically 50-55 days. An Hb A1c proportion of 6.0% or less show good long-term glucose control, while values above 7.0% are elevated. This test is especially useful for diabetics.[23]
The functional magnetic resonance imaging (fMRI) machine may use the signal from oxyhemoglobin as it partially aligns these molecules with the magnetic field. The machine sends a series of magnetic pulses at the participant's head or other body structure, slowly knocking the molecules out of alignment, and a radio wave is emitted when they are back in alignment. The machine can then pick up these signals and use them to make scans, which are cross-sectional maps showing blood flow.
Analogues in non-vertebrate organisms
A variety of oxygen transport and binding proteins exist in organisms throughout the animal and plant kingdoms. Organisms including bacteria, protozoans and fungi all have hemoglobin-like proteins whose known and predicted roles include the reversible binding of gaseous ligands. Since many of these proteins contain globins and the heme moiety (iron in a flat porphyrin support), they are often called hemoglobins, even if their overall tertiary structure is very different from that of vertebrate hemoglobin. In particular, the distinction of “myoglobin” and hemoglobin in lower animals is often impossible, because some of these organisms do not contain muscles. Or, they may have a recognizable separate circulatory system but not one which deals with oxygen transport (for example, many insects and other arthropods). In all these groups, heme/globin containing molecules (even monomeric globin ones) which deal with gas-binding are referred to as hemoglobins. In addition to dealing with transport and sensing of oxygen, they may also deal with NO, CO2, sulfide compounds, and even O2 scavenging in environments which must be anaerobic. They may even deal with detoxification of chlorinated materials in a way analogous to heme-containing P450 enzymes and peroxidases.

The structure of hemoglobins varies across species. Hemoglobin occurs in all kingdoms of organisms, but not in all organisms. Primitive species such as bacteria, protozoa, algae, and plants often have single-globin hemoglobins. Many nematode worms, molluscs and crustaceans contain very large multisubunit molecules, much larger than those in vertebrates. Particularly, chimeric hemoglobins found in fungi and giant annelids may contain both globin and other types of proteins.[24]
One of the most striking occurrences and uses of hemoglobin in organisms is in the giant tube worm (Riftia pachyptila, also called Vestimentifera) which can reach 2.4 meters length and populates ocean volcanic vents. Instead of a digestive tract, these worms contain a population of bacteria constituting half the organism’s weight. The bacteria react with H2S from the vent and O2 from the water to produce energy to make food from H2O and CO2. The worms end with a deep red fan-like structure ("plume") which extends into the water and absorbs H2S and O2 for the bacteria, and CO2 for use as synthetic raw material similar to photosynthetic plants. The structures are bright red due to containing several extraordinarily complex hemoglobins which have up to 144 globin chains, each presumably including associated heme structures. These hemoglobins are remarkable for being able to carry oxygen in the presence of sulfide, and even to carry sulfide, without being completely "poisoned" or inhibited by it as hemoglobins in most other species are.[25][26]
Other oxygen-binding proteins
Myoglobin: Found in the muscle tissue of many vertebrates, including humans, it gives muscle tissue a distinct red or dark gray color. It is very similar to hemoglobin in structure and sequence, but is not a tetramer; instead, it is a monomer that lacks cooperative binding. It is used to store oxygen rather than transport it.
Hemocyanin: The second most common oxygen-transporting protein found in nature, it is found in the blood of many arthropods and molluscs. Uses copper prosthetic groups instead of iron heme groups and is blue in color when oxygenated.
Hemerythrin: Some marine invertebrates and a few species of annelid use this iron-containing non-heme protein to carry oxygen in their blood. Appears pink/violet when oxygenated, clear when not.
Chlorocruorin: Found in many annelids, it is very similar to erythrocruorin, but the heme group is significantly different in structure. Appears green when deoxygenated and red when oxygenated.
Vanabins: Also known as vanadium chromagens, they are found in the blood of sea squirts and are hypothesised to use the rare metal vanadium as its oxygen binding prosthetic group.
Erythrocruorin: Found in many annelids, including earthworms, it is a giant free-floating blood protein containing many dozens — possibly hundreds — of iron- and heme-bearing protein subunits bound together into a single protein complex with a molecular mass greater than 3.5 million daltons.
Pinnaglobin: Only seen in the mollusc Pinna squamosa. Brown manganese-based porphyrin protein.
Leghemoglobin: In leguminous plants, such as alfalfa or soybeans, the nitrogen fixing bacteria in the roots are protected from oxygen by this iron heme containing oxygen-binding protein. The specific enzyme protected is nitrogenase, which is unable to reduce nitrogen gas in the presence of free oxygen.
In history, art and music

Historically, the color of blood was associated with rust, as ancient Romans associated the planet Mars with the god of war since Mars is orange-red. The color of Mars is due to iron-oxygen in the Martian soil, but the red in blood is not due to the iron in hemoglobin and its oxides, which is a common misconception. The red is due to the porphyrin moiety of hemoglobin to which the iron is bound, not the iron itself,[27] although the ligation and redox state of the iron can influence the pi to pi* electronic transitions of the porphyrin and hence its optical characteristics.

Artist Julian Voss-Andreae created a sculpture called "Heart of Steel (Hemoglobin)" in 2005, based on the protein's backbone. The sculpture was made from glass and weathering steel. The intentional rusting of the initially shiny work of art mirrors hemoglobin's fundamental chemical reaction of oxygen binding to iron.[28]
Rock band Placebo recorded a song called Haemoglobin with the lyrics "Haemoglobin is the key to a healthy heartbeat".
See also
- Proteopedia Hemoglobin
- Chlorophyll
- Globin fold
- Hemocyanin
- Hemoprotein
- Sickle-cell disease
Hemoglobin variants:
- Hb A1C
- Hemoglobin A2
- Hemoglobin C
- Hemoglobin F
Hemoglobin protein subunits (genes):
- Alpha globin 1
- Beta globin
- Delta globin
References
- ↑ Maton, Anthea; Jean Hopkins, Charles William McLaughlin, Susan Johnson, Maryanna Quon Warner, David LaHart, Jill D. Wright (1993). Human Biology and Health. Englewood Cliffs, New Jersey, USA: Prentice Hall. ISBN 0-13-981176-1.
- ↑ Funke O (1851). "Uber das milzvenenblut". Z Rat Med 1: 172–218.
- ↑ "A NASA Recipe For Protein Crystallography". Educational Brief. National Aeronautics and Space Administration. Retrieved on 2008-10-12.
- ↑ Hoppe-Seylor F (1866). "Uber die oxydation in lebendem blute". Med-chem Untersuch Lab 1: 133–140.
- ↑ Perutz, M.F.; Rossmann, M.G.; Cullis, A.F.; Muirhead, H.; Will, G.; North, A.C.T. (1960), "Structure of H", Nature 185 (4711): 416–422, doi:
- ↑ Perutz MF (November 1960). "Structure of hemoglobin". Brookhaven symposia in biology 13: 165–83. PMID 13734651.
- ↑ A Syllabus of Human Hemoglobin Variants (1996)
- ↑ Hemoglobin Variants
- ↑ Uthman, MD, Ed. "Hemoglobinopathies and Thalassemias". Retrieved on 2007-12-26.
- ↑ "Hemoglobin Synthesis" (14). Retrieved on 2007-12-26.
- ↑ Steinberg 2001, p. 95
- ↑ Hardison 1996, p. 1
- ↑ Linberg R, Conover CD, Shum KL, Shorr RG (1998). "Hemoglobin based oxygen carriers: how much methemoglobin is too much?". Artif Cells Blood Substit Immobil Biotechnol 26 (2): 133–48. PMID 9564432.
- ↑ Baillie/Simpson. "Online model of the haemoglobin binding and the effects of hyperventilation". Retrieved on 2006-08-10.
- ↑ Childs PE (2001). "Haemoglobin - a molecular lung: 2". Chemistry in Action (65). ISSN 0332-2637. http://www.ul.ie/~childsp/CinA/Issue65/TOC28_Haemoglobin.htm.
- ↑ Rang, H.P.; Dale M.M., Ritter J.M., Moore P.K. (2003). Pharmacology, Fifth Edition. Elsevier. ISBN 04430727027.
- ↑ "Hemoglobin Variants". Lab Tests Online. American Association for Clinical Chemistry (2007-11-10). Retrieved on 2008-10-12.
- ↑ Huisman THJ (1996). "A Syllabus of Human Hemoglobin Variants". Globin Gene Server. Pennsylvania State University. Retrieved on 2008-10-12.
- ↑ Johnson RA, Lavesa M, Askari B, Abraham NG, Nasjletti A (February 1995). "A heme oxygenase product, presumably carbon monoxide, mediates a vasodepressor function in rats". Hypertension 25 (2): 166–9. PMID 7843765. http://hyper.ahajournals.org/cgi/content/abstract/25/2/166. Retrieved on 2008-10-12.
- ↑ Hemoglobin at Medline Plus
- ↑ Hemoglobin Level Test
- ↑ "Hematocrit (HCT) or Packed Cell Volume (PCV)". DoctorsLounge.com. Retrieved on 2007-12-26.
- ↑ This Hb A1c level is only useful in individuals who have red blood cells (RBCs) with normal survivals (i.e., normal half-life). In individuals with abnormal RBCs, whether due to abnormal hemoglobin molecules (such as Hemoglobin S in Sickle Cell Anemia) or RBC membrane defects - or other problems, the RBC half-life is frequently shortened. In these individuals an alternative test called "fructosamine level" can be used. It measures the degree of glycation (glucose binding) to albumin, the most common blood protein, and reflects average blood glucose levels over the previous 18-21 days, which is the half-life of albumin molecules in the circulation.
- ↑ Weber RE, Vinogradov SN (2001). "Nonvertebrate hemoglobins: functions and molecular adaptations". Physiol. Rev. 81 (2): 569–628. PMID 11274340.
- ↑ Zal F, Lallier FH, Green BN, Vinogradov SN, Toulmond A (1996). "The multi-hemoglobin system of the hydrothermal vent tube worm Riftia pachyptila. II. Complete polypeptide chain composition investigated by maximum entropy analysis of mass spectra". J. Biol. Chem. 271 (15): 8875–81. PMID 8621529.
- ↑ Minic Z, Hervé G (2004). "Biochemical and enzymological aspects of the symbiosis between the deep-sea tubeworm Riftia pachyptila and its bacterial endosymbiont". Eur. J. Biochem. 271 (15): 3093–102. doi:. PMID 15265029.
- ↑ Boh, Larry (2001). Pharmacy Practice Manual: A Guide to the Clinical Experience. Lippincott Williams & Wilkins. ISBN 0781725410.
- ↑ Holden, Constance (30 September 2005). "Blood and Steel" (pdf). Science 309: 2160. doi:. http://www.sciencemag.org/cgi/reprint/309/5744/2160d.pdf.
Further reading
|
|
External links
- Interactive models of hemoglobin (Requires MDL Chime)
- Interactive hemoglobin saturation curves
- National Anemia Action Council - anemia.org
- Pulse Oximetry Glossary
- New hemoglobin type causes mock diagnosis with pulse oxymeters
|
||||||||||||||||||||