Internal energy
| Statistical mechanics | ||||||||||||
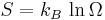 |
||||||||||||
| Statistical thermodynamics Kinetic theory
|
||||||||||||
In thermodynamics, the internal energy of a thermodynamic system, or a body with well-defined boundaries, denoted by U, or sometimes E, is the total of the kinetic energy due to the motion of molecules (translational, rotational, vibrational) and the potential energy associated with the vibrational and electric energy of atoms within molecules or crystals. It includes the energy in all the chemical bonds, and the energy of the free, conduction electrons in metals.
One can also calculate the internal energy of electromagnetic or blackbody radiation. It is a state function of a system, and is an extensive quantity. The SI unit of energy is the joule although other historical, conventional units are still in use, such as the (small and large) calorie for heat.
Contents |
Overview
Internal energy does not include the translational or rotational kinetic energy of a body as a whole. It also does not include the relativistic mass-energy equivalent E = mc2. It excludes any potential energy a body may have because of its location in external gravitational or electrostatic field, although the potential energy it has in a field due to an induced electric or magnetic dipole moment does count, as does the energy of deformation of solids (stress-strain).
The principle of equipartition of energy in classical statistical mechanics states that each molecular quadratic degree of freedom receives 1/2 kT of energy, [1] a result which was modified when quantum mechanics explained certain anomalies; e.g., in the observed specific heats of crystals (when hν > kT). For monoatomic helium and other noble gases, the internal energy consists only of the translational kinetic energy of the individual atoms. Monoatomic particles, of course, do not (sensibly) rotate or vibrate, and are not electronically excited to higher energies except at very high temperatures.
From the standpoint of statistical mechanics, the internal energy is equal to the ensemble average of the total energy of the system.
Composition
Internal energy – the sum of all microscopic forms of energy of a system. It is related to the molecular structure and the degree of molecular activity and may be viewed as the sum of kinetic and potential energies of the molecules; it is composed of the following types of energies:[2]
| Type | Composition of Internal Energy (U) |
|---|---|
| Sensible energy | the portion of the internal energy of a system associated with kinetic energies (molecular translation, rotation, and vibration; electron translation and spin; and nuclear spin) of the molecules. |
| Latent energy | the internal energy associated with the phase of a system. |
| Chemical energy | the internal energy associated with the atomic bonds in a molecule. |
| Nuclear energy | the very large amount of energy associated with the strong bonds within the nucleus of the atom itself. |
| Energy interactions | those types of energies not stored in the system (e.g. heat transfer, mass transfer, and work), but which are recognized at the system boundary as they cross it, which represent gains or losses by a system during a process. |
Sensible energy and latent energy may be further combined into thermal energy.
The first law of thermodynamics
The internal energy is essentially defined by the first law of thermodynamics which states that energy is conserved:
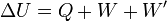
where
- ΔU is the change in internal energy of a system during a process.
- Q is heat added to a system (measured in joules in SI); that is, a positive value for Q represents heat flow into a system while a negative value denotes heat flow out of a system.
- W is the mechanical work done on a system (measured in joules in SI)
- W' is energy added by all other processes
The first law may be stated equivalently in infinitesimal terms as:
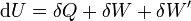
where the terms now represent infinitesimal amounts of the respective quantities. The d before the internal energy function indicates that it is an exact differential. In other words it is a state function or a value which can be assigned to the system. On the other hand, the δ's before the other terms indicate that they describe increments of energy which are not state functions but rather they are processes by which the internal energy is changed. (See the discussion in the first law article.)
From a microscopic point of view, the internal energy may be found in many different forms. For a gas it may consist almost entirely of the kinetic energy of the gas molecules. It may also consist of the potential energy of these molecules in a gravitational, electric, or magnetic field. For any material, solid, liquid or gaseous, it may also consist of the potential energy of attraction or repulsion between the individual molecules of the material.
Expressions for the internal energy
The internal energy may be expressed in terms of other thermodynamic parameters. Each term is composed of an intensive variable (a generalized force) and its conjugate infinitesimal extensive variable (a generalized displacement).
For example, for a non-viscous fluid, the mechanical work done on the system may be related to the pressure p and volume V. The pressure is the intensive generalized force, while the volume is the extensive generalized displacement:
Taking the default direction of work, 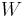 , to be from the working fluid to the surroundings,
, to be from the working fluid to the surroundings,
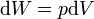 .
.
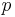 is the
is the 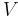 is the
is the Taking the default direction of heat transfer, 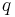 , to be into the working fluid and assuming a reversible process, we have
, to be into the working fluid and assuming a reversible process, we have
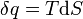 .
.
 is temperature
is temperature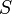 is entropy
is entropy
The above two equations in the first law of thermodynamics imply for a closed system:
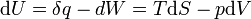
If we also include the dependence on the numbers of particles in the system, the internal energy function may be written as 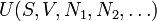 where the
where the 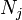 are the numbers of particles of type j in the system. The fact that U is an extensive function when considered as a function of the variables S, V,
are the numbers of particles of type j in the system. The fact that U is an extensive function when considered as a function of the variables S, V, 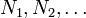 , we have:
, we have:
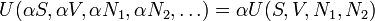
From Euler's homogeneous function theorem we may now write the internal energy as:
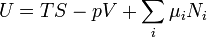
where the 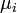 are the chemical potentials for the particles of type i in the system. These are defined as:
are the chemical potentials for the particles of type i in the system. These are defined as:
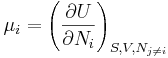
For an elastic substance the mechanical term must be replaced by the more general expression involving the stress 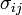 and strain
and strain 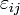 . The infinitesimal statement is:
. The infinitesimal statement is:
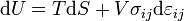
where Einstein notation has been used for the tensors, in which there is a summation over all repeated indices in the product term. The Euler theorem yields for the internal energy (Landau & Lifshitz 1986):
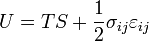
For a linearly elastic material, the stress can be related to the strain by:
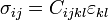
Change in internal energy due to change in temperature and volume or pressure
The expressions given above for the internal energy involves the entropy. In practice one often wants to know the change in internal energy of a substance as a function of the change in temperature and volume, or as a function of the change in temperature and pressure.
To express dU in terms of dT and dV, we substitute
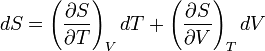
in the fundamental thermodynamic relation
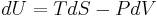
This gives:
![dU = T\left(\frac{\partial S}{\partial T}\right)_{V}dT +\left[T\left(\frac{\partial S}{\partial V}\right)_{T} - P\right]dV\,](/2009-wikipedia_en_wp1-0.7_2009-05/I/687594e325ce89f0e8ce28b9627f6767.png)
The term 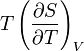 is the heat capacity at constant volume
is the heat capacity at constant volume 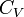 .
.
The partial derivative of S w.r.t. V can be evaluated if the equation of state is known. From the fundamental thermodynamic relation, it follows that the differential of the Helmholtz free energy F is given by:
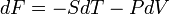
The symmetry of second derivatives of F w.r.t. T and V yields the Maxwell relation:
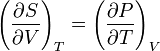
This gives the expression:
![dU =C_{V}dT +\left[T\left(\frac{\partial P}{\partial T}\right)_{V} - P\right]dV\,\,\text{ (1)}\,](/2009-wikipedia_en_wp1-0.7_2009-05/I/e59f045309ad50d93824497a9556ef11.png)
This is useful if the equation of state is known. In case of an ideal gas, 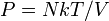 which implies that
which implies that 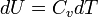 , i.e. the internal energy of an ideal gas can be written as a function that depends only on the temperature.
, i.e. the internal energy of an ideal gas can be written as a function that depends only on the temperature.
When dealing with fluids or solids, an expression in terms of the temperature and pressure is usually more useful. The partial derivative of the pressure w.r.t. temperature at constant volume can be expressed in terms of the coefficient of thermal expansion
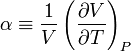
and the isothermal compressibility
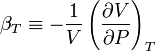
by writing:
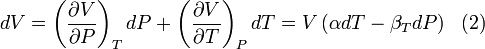
and equating dV to zero and solving for the ratio dP/dT. This gives:
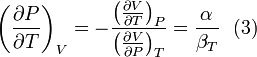
Substituting (2) and (3) in (1) gives:
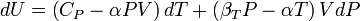
where we have used that the heat capacity at constant pressure is related to the heat capacity at constant volume according to:
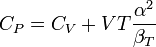
as shown here.
References
- Alberty, R. A. (2001). "Use of Legendre transforms in chemical thermodynamics" (PDF). Pure Appl. Chem. Vol. 73 (8): 1349–1380. doi:. http://www.iupac.org/publications/pac/2001/pdf/7308x1349.pdf.
- Lewis, Gilbert Newton; Randall, Merle: Revised by Pitzer, Kenneth S. & Brewer, Leo (1961). Thermodynamics (2nd Edition ed.). New York, NY USA: McGraw-Hill Book Co.. ISBN 0-07-113809-9.
- Landau, L. D.; Lifshitz, E. M. (1986). Theory of Elasticity (Course of Theoretical Physics Volume 7). (Translated from Russian by J.B. Sykes and W.H. Reid) (Third ed. ed.). Boston, MA: Butterworth Heinemann. ISBN 0-7506-2633-X.
Notes
See also
- Calorimetry
- Thermodynamic equations
- Thermodynamic potentials
- Gibbs free energy