Cell cycle
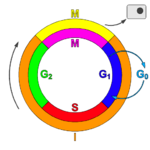
The cell cycle, or cell-division cycle, is the series of events that take place in a cell leading to its replication. In prokaryotes, the cell cycle occurs via a process termed binary fission. In eukaryotes, the cell cycle can be divided in two brief periods: interphase—during which the cell grows, accumulating nutrients needed for mitosis and duplicating its DNA—and the mitotic (M) phase, during which the cell splits itself into two distinct cells, often called "daughter cells". The cell-division cycle is a vital process by which a single-celled fertilized egg develops into a mature organism, as well as the process by which hair, skin, blood cells, and some internal organs are renewed.
Contents |
Regulation of eukaryotic cell cycle

Regulation of the cell cycle involves processes crucial to the survival of a cell, including the detection and repair of genetic damage as well as the prevention of uncontrolled cell division. The molecular events that control the cell cycle are ordered and directional; that is, each process occurs in a sequential fashion and it is impossible to "reverse" the cycle.
Role of Cyclins and CDKs
Two key classes of regulatory molecules, cyclins and cyclin-dependent kinases (CDKs), determine a cell's progress through the cell cycle.[1] Leland H. Hartwell, R. Timothy Hunt, and Paul M. Nurse won the 2001 Nobel Prize in Physiology or Medicine for their discovery of these central molecules.[2] Many of the genes encoding cyclins and CDKs are conserved among all eukaryotes, but in general more complex organisms have more elaborate cell cycle control systems that incorporate more individual components. Many of the relevant genes were first identified by studying yeast, especially Saccharomyces cerevisiae;[3] genetic nomenclature in yeast dubs many these genes cdc (for "cell division cycle") followed by an identifying number, e.g., cdc25.
Cyclins form the regulatory subunits and CDKs the catalytic subunits of an activated heterodimer; cyclins have no catalytic activity and CDKs are inactive in the absence of a partner cyclin. When activated by a bound cyclin, CDKs perform a common biochemical reaction called phosphorylation that activates or inactivates target proteins to orchestrate coordinated entry into the next phase of the cell cycle. Different cyclin-CDK combinations determine the downstream proteins targeted. CDKs are constitutively expressed in cells whereas cyclins are synthesised at specific stages of the cell cycle, in response to various molecular signals.[4]
General mechanism of cyclin-CDK interaction
Upon receiving a pro-mitotic extracellular signal, G1 cyclin-CDK complexes become active to prepare the cell for S phase, promoting the expression of transcription factors that in turn promote the expression of S cyclins and of enzymes required for DNA replication. The G1 cyclin-CDK complexes also promote the degradation of molecules that function as S phase inhibitors by targeting them for ubiquitination. Once a protein has been ubiquitinated, it is targeted for proteolytic degradation by the proteasome. Active S cyclin-CDK complexes phosphorylate proteins that make up the pre-replication complexes assembled during G1 phase on DNA replication origins. The phosphorylation serves two purposes: to activate each already-assembled pre-replication complex, and to prevent new complexes from forming. This ensures that every portion of the cell's genome will be replicated once and only once. The reason for prevention of gaps in replication is fairly clear, because daughter cells that are missing all or part of crucial genes will die. However, for reasons related to gene copy number effects, possession of extra copies of certain genes would also prove deleterious to the daughter cells.
Mitotic cyclin-CDK complexes, which are synthesized but inactivated during S and G2 phases, promote the initiation of mitosis by stimulating downstream proteins involved in chromosome condensation and mitotic spindle assembly. A critical complex activated during this process is a ubiquitin ligase known as the anaphase-promoting complex (APC), which promotes degradation of structural proteins associated with the chromosomal kinetochore. APC also targets the mitotic cyclins for degradation, ensuring that telophase and cytokinesis can proceed.
Specific action of cyclin-CDK complexes
Cyclin D is the first cyclin produced in the cell cycle, in response to extracellular signals (eg. growth factors). Cyclin D binds to existing CDK4, forming the active cyclin D-CDK4 complex. Cyclin D-CDK4 complex in turn phosphorylates the retinoblastoma susceptibility protein (RB). The hyperphosphorylated RB dissociates from the E2F/DP1/RB complex (which was bound to the E2F responsive genes, effectively "blocking" them from transcription), activating E2F. Activation of E2F results in transcription of various genes like cyclin E, cyclin A, DNA polymerase, thymidine kinase, etc. Cyclin E thus produced binds to CDK2, forming the cyclin E-CDK2 complex, which pushes the cell from G1 to S phase (G1/S transition). Cyclin B along with cdc2 forms the cyclin B-cdc2 complex, which initiates the G2/M transition.[5] Cyclin B-cdc2 complex activation causes breakdown of nuclear envelope and initiation of prophase, and subsequently, its deactivation causes the cell to exit mitosis.[4]
Cell cycle inhibitors
Two families of genes, the cip/kip family and the INK4a/ARF (Inhibitor of Kinase 4/Alternative Reading Frame) prevent the progression of the cell cycle. Because these genes are instrumental in prevention of tumor formation, they are known as tumor suppressors.
The cip/kip family includes the genes p21, p27 and p57. They halt cell cycle in G1 phase, by binding to, and inactivating, cyclin-CDK complexes. p21 is activated by p53 (which, in turn, is triggered by DNA damage eg. due to radiation). p27 is activated by Transforming Growth Factor β (TGF β), a growth inhibitor.
The INK4a/ARF family includes p16INK4a, which binds to CDK4 and arrests the cell cycle in G1 phase, and p14arf which prevents p53 degradation. And the amount of chromosomes are able to double at the same rate as in phase 2
Checkpoints
Cell cycle checkpoints are used by the cell to monitor and regulate the progress of the cell cycle.[6] Checkpoints prevent cell cycle progression at specific points, allowing verification of necessary phase processes and repair of DNA damage. The cell cannot proceed to the next phase until checkpoint requirements have been met.
Several checkpoints are designed to ensure that damaged or incomplete DNA is not passed on to daughter cells. Two main checkpoints exist: the G1/S checkpoint and the G2/M checkpoint. G1/S transition is a rate-limiting step in the cell cycle and is also known as restriction point.[4] An alternative model of the cell cycle response to DNA damage has also been proposed, known as the postreplication checkpoint.
p53 plays an important role in triggering the control mechanisms at both G1/S and G2/M checkpoints.
Miscellaneous information
Role of cell cycle in tumor formation
A disregulation of the cell cycle components may lead to tumor formation. As mentioned above, some genes like the cell cycle inhibitors, RB, p53 etc., when they mutate, may cause the cell to multiply uncontrollably, forming a tumor. Although the duration of cell cycle in tumor cells is equal to or longer than that of normal cell cycle, the proportion of cells that are in active cell division (versus quiescent cells in G0 phase) in tumor cells are much more compared to that in normal cells. Thus there is a net increase in cell number as the number of cells that die by apoptosis or senescence remains the same.
The cells which are actively undergoing cell cycle are targeted in cancer therapy as the DNA is relatively exposed during cell division and hence susceptible to damage by drugs or radiation. This fact is made use of in cancer treatment; by a process known as debulking, a significant mass of the tumor is removed which pushes a significant number of the remaining tumor cells from G0 to G1 phase (due to increased availability of nutrients, oxygen, growth factors etc.). Radiation or chemotherapy following the debulking procedure kills these cells which have newly entered the cell cycle. [4]
Synchronization of cell cultures
Several methods can be used to synchronise cell cultures by halting the cell cycle at a particular phase. For example, Serum starvation [7] and treatment with Thymidine or Aphidicolin [8] halt the cell in the G1 phase, Mitotic shake-off, treatment with colchicine [9] and treatment with Nocodazole [10] halt the cell in M phase and treatment with 5-fluorodeoxyuridine halts the cell in S phase.
Observation
There are numerous ways to observe the cell cycle occurring. Onion bulbs or garlic root tips are often used.
A sample of root tip is fixed in a mixture of 99% of 70% aqueous industrial methylated spirit and 1% glacial ethanoic acid for two hours. Treat the root tips in 1 molar hydrochloric acid at 60°C for 6–7 minutes. Rinse thoroughly with water. Add Schiff's reagent and leave for one hour. Rinse again in distilled water. Observe under a microscope.
Mathematical modelling
See cell cycle mathematical model
See also
- Topic outline of cell biology
- Mitosis
- Interphase
References
- ↑ "Cyclin-dependent protein kinases: key regulators of the eukaryotic cell cycle". Bioessays 17 (6): 471-80. June 1995. doi:. PMID 7575488. http://www.ncbi.nlm.nih.gov/sites/entrez?cmd=Retrieve&db=PubMed&list_uids=7575488&dopt=Citation.
- ↑ "Press release". Nobelprize.org.
- ↑ "Comprehensive Identification of Cell Cycle-regulated Genes of the Yeast Saccharomyces cerevisiae by Microarray Hybridization". Molecular Biology of the Cell 9: 3273-3297. December 1998. PMID 9843569. http://www.molbiolcell.org/cgi/content/abstract/9/12/3273.
- ↑ 4.0 4.1 4.2 4.3 Robbins and Cotran; Kumar, Abbas, Fausto (2004). Pathological Basis of Disease. Elsevier. ISBN 81-8147-528-3.
- ↑ Norbury, C. (1995) "Cdc2 protein kinase (vertebrates). In:The Protein Kinase Facts Book: Protein-Serine Kinases(Hardie, G. and Hanks, S., eds.), Academic Press. 184.
- ↑ Stephen J. Elledge (6 December 1996). "Cell Cycle Checkpoints: Preventing an Identity Crisis". Science 274 (5293): 1664-1672. doi:. PMID 8939848. http://www.sciencemag.org/cgi/content/abstract/274/5293/1664.
- ↑ "Cell Cycle Synchronization of Porcine Fetal Fibroblasts: Effects of Serum Deprivation and Reversible Cell Cycle Inhibitors". Biology of Reproduction 62: 412-419. 2000. doi:. PMID 10642581. http://www.biolreprod.org/cgi/content/abstract/62/2/412.
- ↑ G Pedrali-Noy, S Spadari, A Miller-Faurès, A O Miller, J Kruppa, and G Koch (1980 January 25;). "Synchronization of HeLa cell cultures by inhibition of DNA polymerase alpha with aphidicolin". Nucleic Acids Res 8 (2): 377–387. doi:. PMID 6775308. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=327273.
- ↑ R.S. Prather, A.C. Boquest, B.N. Day (1999). "Cell Cycle Analysis of Cultured Porcine Mammary Cells". Cloning 1 (1): 17-24. doi:. PMID 16218827. http://www.liebertonline.com/doi/abs/10.1089/15204559950020067?cookieSet=1&journalCode=clo.
- ↑ Seydou Samaké, Lawrence C. Smith (1997-10-15). "Synchronization of cell division in eight-cell bovine embryos produced in vitro: Effects of nocodazole". Theriogenology 48 (6): 969-76. doi:. PMID 16728186.
- Morgan DO. (2007) The Cell Cycle: Principles of Control. New Science Press: London.
- Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. (2003). Molecular Biology of the Cell. Ch 17. Garland Science: New York. 4th ed.
- Lodish H, Berk A, Matsudaira P, Kaiser CA, Krieger M, Scott MP, Zipursky SL, Darnell J. (2004). Molecular Cell Biology. WH Freeman: New York, NY. 5th ed.
- Watson JD, Baker TA, Bell SP, Gann A, Levine M, Losick R. (2004). Molecular Biology of the Gene, Ch. 7. Peason Benjamin Cummings; CSHL Press. 5th ed.
External links
- This article contains material from the Science Primer published by the NCBI, which, as a U.S. government publication, is in the public domain.
- Transcriptional program of the cell cycle: high-resolution timing
- Cell cycle and metabolic cycle regulated transcription in yeast
- Cell Cycle Animation 1Lec.com
- Cell Cycle and Cytokinesis - The Virtual Library of Biochemistry and Cell Biology
- Cell Cycle
- Cell Cycle Portal
- Fucci:Using GFP to visualize the cell-cycle
- Science Creative Quarterly's overview of the cell cycle
- Cells alive
- CCO The Cell-Cycle Ontology
- KEGG - Human Cell Cycle
- Cell cycle modeling
|
||||||||||||||||
|
||||||||||||||